
Download
ORIGINAL ARTICLE
CTRP3 regulates NF-κB and TGFβ1/Smad3 pathways to alleviate airway inflammation and remodeling in asthmatic mice induced by OVA
Hai Lina, Jinrong Yib*
aDepartment of Respiratory and Critical Care Medicine, First Affiliated Hospital of Gannan Medical University, Ganzhou, Jiangxi, China
bDepartment of Anesthesiology, Ganzhou Women and Children’s Health Care Hospital, Ganzhou, Jiangxi, China
Abstract
Background: Asthma is a common illness with chronic airway inflammation. C1q/tumor necrosis factor (TNF)-related protein 3 (CTRP3) plays a vital role ininflammatory response, but its effect on asthma is imprecise. Herein, we analyzed the functions of CTRP3 in asthma.
Methods: The BALB/c mice were randomized into four groups: control, ovalbumin (OVA), OVA+vector, and OVA+CTRP3. The asthmatic mice model was established by OVA stimulation. Overexpression of CTRP3 was implemented by the transfection of corresponding adeno-associated virus 6 (AAV6). The contents of CTRP3, E-cadherin, N-cadherin, smooth muscle alpha-actin (α-SMA), phosphorylated (p)-p65/p65, transforming growth factor-beta 1 (TGFβ1), and p-Smad3/Smad3 were determined by Western blot analysis. The quantity of total cells, eosinophils, neutrophils, and lymphocytes in bronchoalveolar lavage fluid (BALF) was assessed by using a hemocytometer. The contents of tumor necrosis factor-α and interleukin-1β in BALF were examined by enzyme-linked immunesorbent serologic assay. The lung function indicators and airway resistance (AWR) were measured. The bronchial and alveolar structures were evaluated by hematoxylin and eosin staining and sirius red staining.
Results: The CTRP3 was downregulated in mice of OVA groups; however, AAV6-CTRP3 treatment markedly upregulated the expression of CTRP3. Upregulation of CTRP3 diminished asthmatic airway inflammation by decreasing the number of inflammatory cells and the contents of proinflammatory factors. CTRP3 markedly lessened AWR and improved lung function in OVA-stimulated mice. Histological analysis found that CTRP3 alleviated OVA-induced airway remodeling in mice. Moreover, CTRP3 modulated NF-κB and TGFβ1/Smad3 pathways in OVA-stimulated mice.
Conclusion: CTRP3 alleviated airway inflammation and remodeling in OVA-induced asthmatic mice via regulating NF-κB and TGFβ1/Smad3 pathways.
Key words: Asthma, airway inflammation, airway remodeling, CTRP3, NF-κB, TGFβ1/Smad3
*Corresponding author: Jinrong Yi, Department of Anesthesiology, Ganzhou Women and Children’s Health Care Hospital, No. 106, Dagong Road, Zhanggong District, Ganzhou, Jiangxi 341000, China. Email address: [email protected]
Received 13 March 2023; Accepted 17 April 2023; Available online 1 July 2023
Copyright: H Lin and J Yi
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Asthma is a heterogeneous chronic inflammatory disease characterized by reversible airflow obstruction. The airway inflammation is the main determinant of airway hyper-responsiveness, which can be triggered by multiple factors, including allergens, infections, and pollutants.1,2 Asthma is one of the most common chronic diseases in children.3 According to the data released by World Health Organization (WHO), about 130 million children suffer from asthma globally.4 According to the WHO estimates, 38,300 people die annually from asthma attacks globally.5 Its increasing prevalence imposes a heavy burden on public health. Asthma is closely associated with the overproduction of various inflammatory mediators, cytokines, and adhesion molecules.6 Airway inflammation and remodeling often occur in asthma patients, leading to histological changes in airway structure, resulting in increased airway fibrosis and decreased lung function.7 Airway inflammation in asthma has been found to be regulated by several factors, including inflammatory cells and various cytokines.8 In the current clinical works, corticosteroids are often used to control asthma but cannot lessen airway structural damage, and their therapeutic effect is limited.9 Therefore, we urgently need new strategies for treating asthma.
C1q/tumor necrosis factor (TNF)-related protein 3 (CTRP3) belongs to a highly conserved CTRP adiponectin superfamily para-homology.10 In adult mice, CTRP3 is considered as an adipokine because it is mainly expressed in adipose tissue. However, CTRP3 is also expressed in the heart, lung, kidney, spleen, testis, and other organs.11 Previous studies have shown that CTRP3 is an endogenous antagonist of lipopolysaccharide, and the abnormal expression of CTRP3 is linked with numerous types of diseases, such as sepsis, myocardial dysfunction, and severe pancreatitis.12,13 CTRP3 also can attenuate TGF-β1-induced Smad3 phosphorylation and improve cardiac fibrosis.14 However, few studies have studied the role of CTRP3 in asthma, and its related regulatory mechanism is not clear.
Herein, we built an asthma mouse model by ovalbumin (OVA) treatment. Besides, we confirmed the role of CTRP3 in airway inflammation and remodeling in asthmatic mice. The purpose of this study was to investigate the role of CTRP3 in asthma and its regulatory molecular mechanism. Our research provided an experimental basis for the improvement of targeted therapy for asthma.
Materials and Methods
Animals
Female BALB/c mice (specific pathogen-free, 6-week old) were obtained from Shanghai Laboratory Animal Company (SLAC, Shanghai, China), and kept in pathogen-free conditions. The animal experiments were conducted according to the guidelines provided in the Guide for the Care and Use of Laboratory Animals.15 These mice were adaptively fed for 1 week with OVA-free food and water. The animal research was approved by the Animal Ethical and Welfare Committee of First Affiliated Hospital of Gannan Medical University.
Creation of OVA-induced mouse model
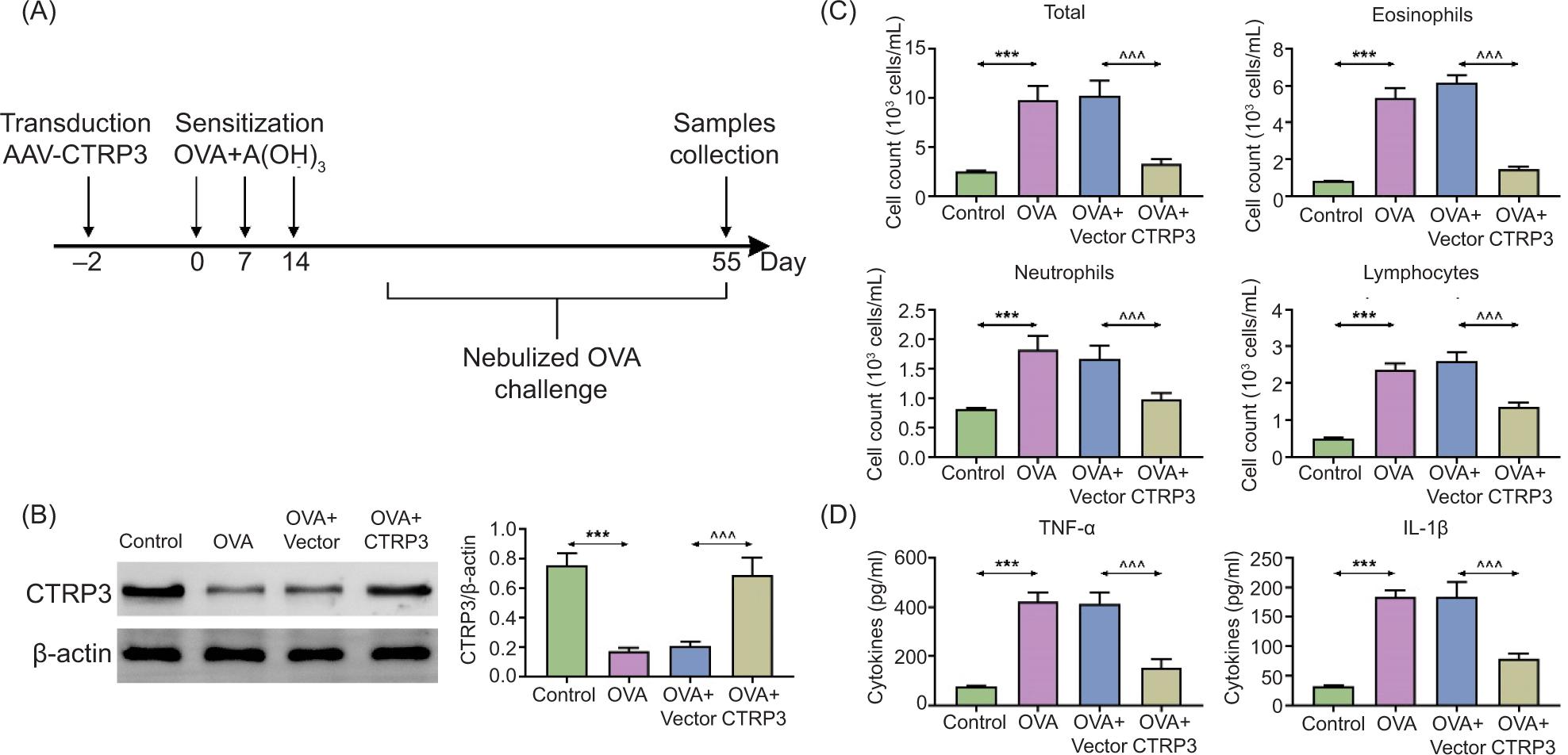
The mice were randomly divided into the following four groups (n = 6 mice/group): control, OVA, OVA+vector, OVA+CTRP3. Mice were sensitized and challenged as reported previously.8 In brief, except for the control group, the mice were sensitized on day 0, 7, and 14 by intraperitoneal (i.p.) injection of OVA (40 μg; Sigma-Aldrich, St. Louis, MO, USA) and 10% Al(OH)3 (2 mg; Sigma-Aldrich) in phosphate-buffered saline solution (PBS; total volume: 0.2 mL; Sigma-Aldrich). The mice were challenged for 20 min through airway with aerosolized 5% OVA in PBS (from day 21 to 55) by utilizing an ultrasonic nebulizer (403 M; YUWELL, Zhenjiang, China). The control group mice were provided the identical management with PBS as a substitute for OVA. The sensitization protocol is shown in Figure 1A.
Figure 1 Upregulation of CTRP3 diminished inflammation of asthmatic airway in OVA-stimulated mice. The BALB/c mice were randomly divided into four groups: control, OVA, OVA+vector, and OVA+CTRP3. (A) The sensitization flow chart of mice. (B) The content of CTRP3 in lung tissues was examined by Western blot analysis. (C) The quantity of total cells, including eosinophils, neutrophils, and lymphocytes, in BALF was assessed by utilizing a hemocytometer. (D) The contents of TNF-α and IL-1β in BALF were examined by ELISA. Compared to the control group, ***P < 0.001; compared to the OVA+vector group, ^^^P < 0.001. All data were expressed as mean ± SD.
Overexpression of CTRP3
Recombinant adeno-associated virus 6 (AAV6) with intense affinity for lung tissues act as a vector.16,17 Recombinant AAV6 (1 × 1012 vg/mL) was constructed by GenePharma (Shanghai, China), comprising green fluorescent protein (GFP) labeling and sequence of interest. The sequence of interest was the control sequence (vector) or the overexpression sequence of CTRP3. The mice of OVA+vector and OVA+CTRP3 groups were transfected 2 days before the first sensitization with corresponding AAV6 to express the interested sequence. After the mice were completely anesthetized with 4% chloral hydrate (0.1 mg/10 g; Sigma-Aldrich) via i.p. injection, 50-μL liquid comprising AAV6 was infused in the lungs by endotracheal intubation.
Western blot analysis
Proteins were collected from the lung tissues of mice, and were separated by using sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE). The proteins were transferred to polyvinylidene fluoride (PVDF) membranes (Sigma-Aldrich). After blocking, the membranes were incubated with primary antibodies overnight at 4°C. Then the membranes were incubated with goat anti-rabbit Immunoglobulin G (IgG, ab205718, 1:2500; Abcam, Cambridge, MA, USA) for 1 h. The membranes were evaluated with a Western blot (WB) kit (Sigma-Aldrich) via a Bio-Rad gel imaging system (Bio-Rad, Hercules, CA, USA). The images were analyzed with the Image J software as described by Rahmati et al.18,19 The primary antibodies included the following: anti-CTRP3 (ab36870, 1:1000; Abcam), anti-E-cadherin (ab76319, 1:1000; Abcam), anti-N-cadherin (ab76011, 1:1000; Abcam), anti-smooth muscle alpha-actin (anti-α-SMA, ab5694, 1:1000; Abcam), anti-phosphorylated (p)-p65 (3033, 1:1000; CST, Danvers, MA, USA), anti-p65 (8242, 1:1000; CST), anti-TGFβ1 (3711, 1:1000; GST), anti-p-Smad3 (9520, 1:1000; Abcam), anti-Smad3 (9531, 1:1000; Abcam), and anti-β-actin (ab8226, 1:1000; Abcam).
Collection and investigation of bronchoalveolar lavage fluid (BALF)
Airway inflammatory response was evaluated at 24 h following the last challenge. BALF was reaped through conveying 0.8-mL cold PBS (Sigma-Aldrich) by endotracheal intubation and lightly suctioning the fluid. The lung lavage was done for three times, with the recovery ratio of about 80%. Then the BALF was centrifuged and cell precipitation and supernatant were collected separately. The supernatant was preserved at -80°C for subsequent analysis. The cell precipitation was resuspended in 1-mL PBS (Sigma-Aldrich), and the quantity of total cells, including eosinophils, neutrophils, and lymphocytes, was assessed by a hemocytometer. The contents of TNF-α and interleukin (IL)-1β in BALF supernatant were examined by exploiting corresponding enzyme-linked immunosorbent serologic assay (ELISA; ab183218 and ab7632; Abcam) kits according to the instructions provided for specific experimental procedures.
Detection of airway resistance (AWR) and lung function
Airway resistance was evaluated at 24 h following the last challenge. Mice were anesthetized with sodium pentobarbital (50 mg/kg; Sigma-Aldrich). Tracheal intubation was performed by using 22G indwelling needle. After hooking up to the mouse lung function instrument, the mice were positioned on the heating plate of a closed incubator. A respirator was utilized for aided respiration. The mice received intravenous (i.v.) infusion of methacholine (12.5, 25, and 50 mg/mL; Sigma-Aldrich). Alterations in AWR (cm H2O s/L), lung function indicators (peak expiratory flow [PEF, mL/s], and the ratio of forced expiratory volume in 0.4 s to forced vitial capacity [forced expiratory volume or FEV0.4/forced vital capacity or FVC]), and respiratory rate (breaths/min) were examined. The AWR in draft PBS was Rbl (baseline). AWR of indrawing methacholine at diverse dosages was Rres (response). The dominant value of AWR at every dosage was recorded and transformed into the fold increase during PBS provocation (R) as an indicator to evaluate AWR according to the following formula:
Fold Increase of R = [Rres (response) – Rbl (baseline)] /Rbl (baseline).
Hematoxylin and eosin (H&E) staining
The lung tissues of mice were dehydrated (gradient ethanol; Sigma-Aldrich), permeabilized, embedded in paraffin (Sigma-Aldrich), and cut into slices. The specific experimental steps are consistent with that of Bostani et al. and Rahmati et al.20,21 The slices were exposed to hematoxylin (Sigma-Aldrich) for 5 min, dealt with ethanol hydrochloride (Sigma-Aldrich) for 3 s, and treated with eosin (Sigma-Aldrich) for 2 min. The slices were observed under a light microscope (Leica, Wetzlar, Germany).
Sirius red staining
For histological analysis, the lung tissues of mice were fixed in 4% paraformaldehyde (PFA; Sigma-Aldrich) and embedded in paraffin (Sigma-Aldrich). The samples were cut into 5-µm slices and stained with sirius red (Sigma-Aldrich). The images were taken by an inverted microscope (Leica).
Statistical assay
All investigations were repeated for more than three times. Statistical analyses were executed using SPSS 22.0 (SPSS, Inc., Chicago, IL, USA). All data were expressed as mean ± SD. Shapiro–Wilk test was used to detect normal distribution. Bartlett’s test was used to detect variance homogeneity. Paired comparison was done using Student’s t-test. Multiple comparison was done by analysis of variance (ANOVA). P < 0.05 was considered as statistically significant.
Results
CTRP3 alleviated inflammation in BALF of OVA-stimulated mice
To inspect the influence of CTRP3 in asthma, we built a mouse model through OVA treatment. The mice were randomly divided into the following four groups: control, OVA, OVA+vector, OVA+CTRP3. We found that the content of CTRP3 in lung tissues was apparently reduced in mice of OVA groups, compared to the control group. After transfection with AAV6–CTRP3, the content of CTRP3 was evidently boosted with respect to the OVA+vector group, suggesting that AAV6–CTRP3 markedly upregulated the expression of CTRP3 in lung tissues (Figure 1B). Besides, we discovered the quantity of total cells, including eosinophils, neutrophils, and lymphocytes (Figure 1C). Besides, the contents of TNF-α and IL-1β (Figure 1D) in BALF were increased by OVA induction, which was lessened by AAV6–CTRP3 co-treatment. Therefore, upregulation of CTRP3 diminished asthmatic airway inflammation by decreasing the number of inflammatory cells and the contents of proinflammatory factors.
CTRP3 lessened AWR in OVA mice
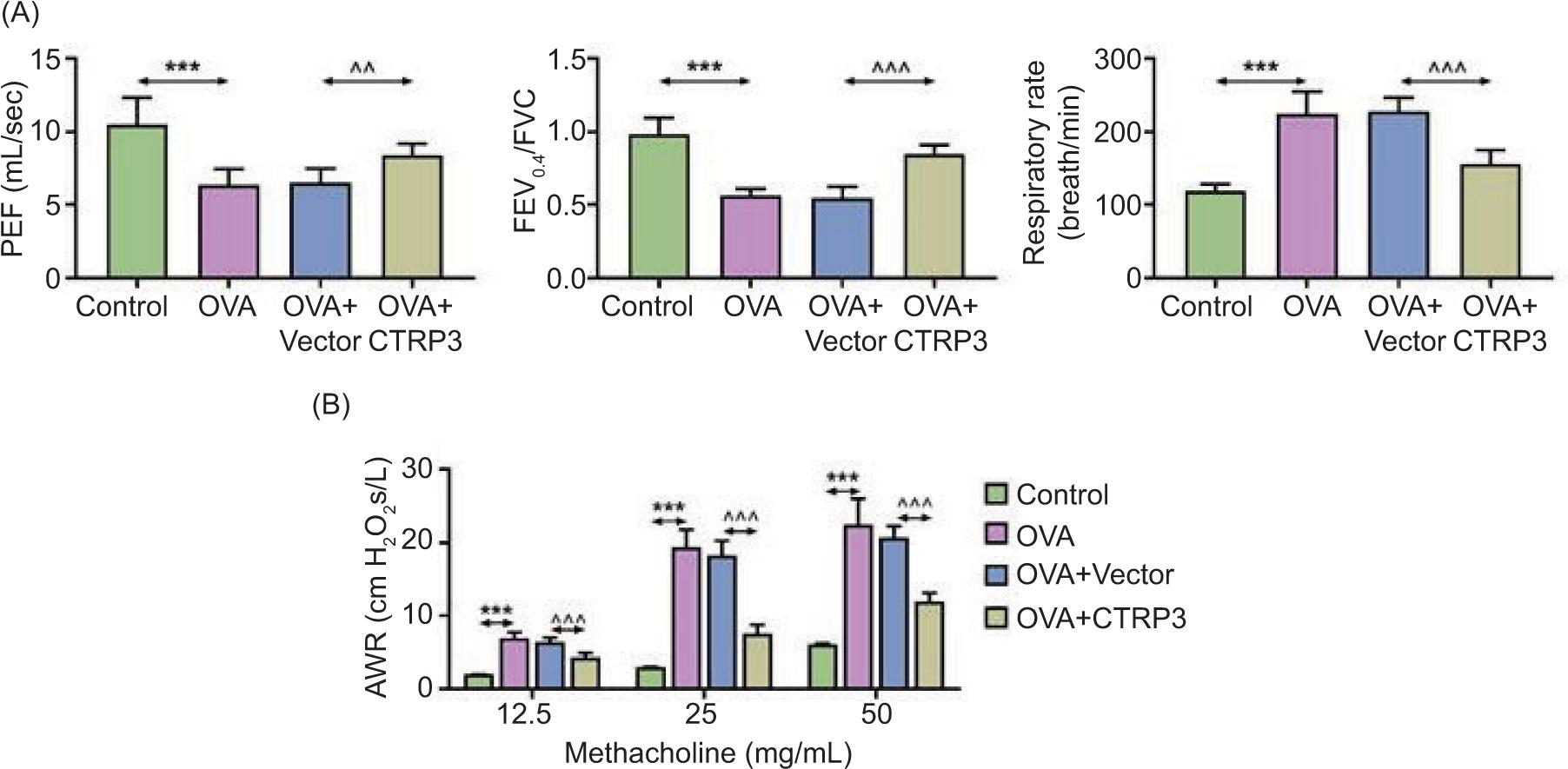
We scrutinized the influence of CTRP3 on the lung function of asthmatic mice. We discovered that lung function indicators, such as PEF and FEV0.4/FVC, declined but respiratory rate was augmented in the OVA groups, compared to the control group. PEF and FEV0.4/FVC were augmented and respiratory rate was diminished in the OVA+CTRP3 group with respect to the OVA+vector group (Figure 2A). Administration of diverse doses of methacholine apparently boosted AWR in the OVA groups but was obviously diminished in the OVA+CTRP3 group (Figure 2B). Hence, we revealed that CTRP3 could markedly lessen AWR and increase lung function in OVA-stimulated mice.
Figure 2 CTRP3 lessened AWR in OVA mice. (A) The lung function indicators, such as PEF, FEV0.4/FVC, and respiratory rate, were assessed in mice. (B) AWR was measured using different doses of methacholine. Compared to the control group, ***P < 0.001; compared to the OVA+vector group, ^^P < 0.01, ^^^P < 0.001. All data were expressed as mean ± SD.
CTRP3 curbed OVA-induced airway remodeling
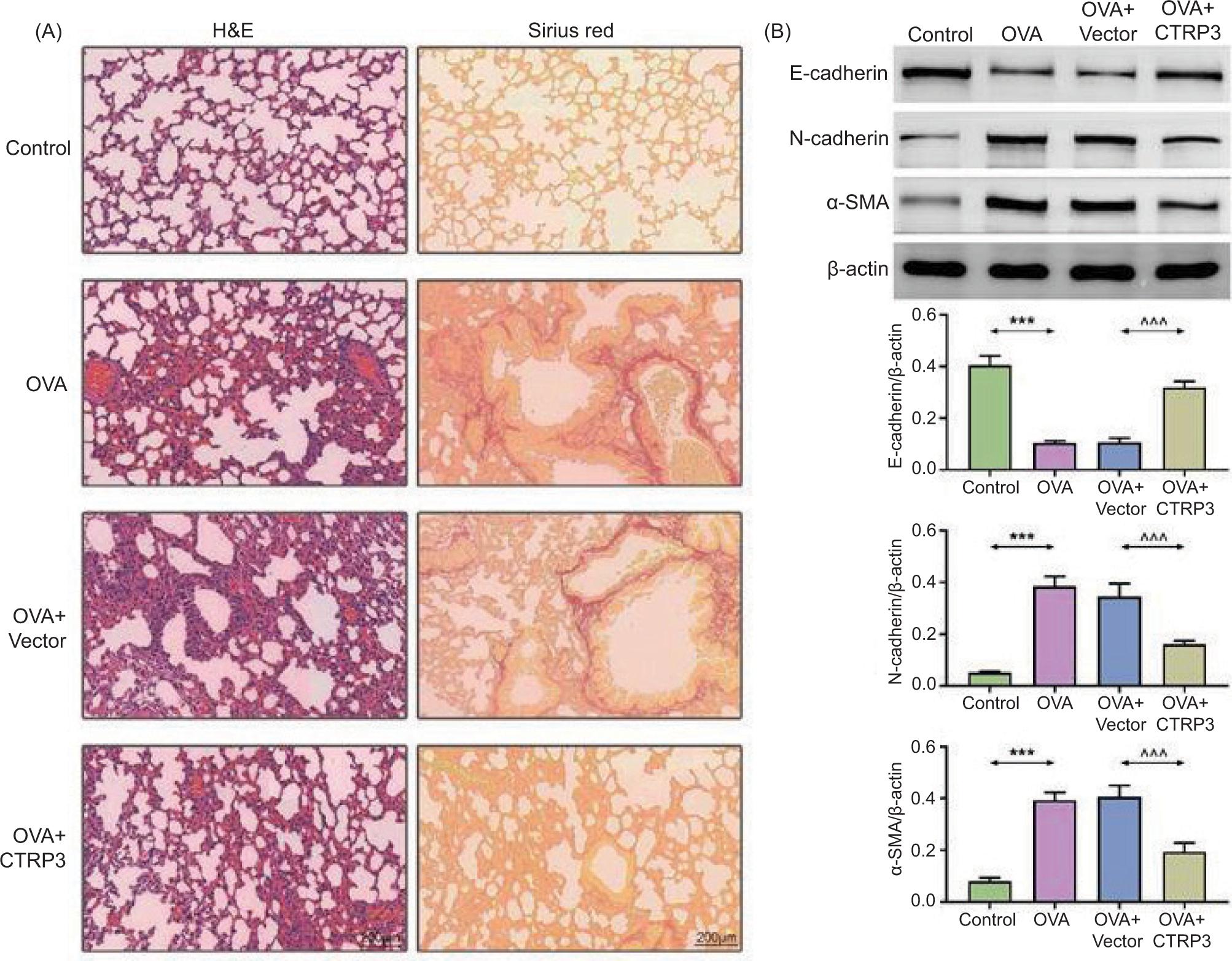
We analyzed the influence of CTRP3 on airway remodeling of asthmatic mice. H&E staining and sirius red staining results revealed that bronchial and alveolar structures were muddled and missing in the OVA groups. The infiltration of inflammatory cells around the bronchial mucosa was severe. The inflammatory cell infiltration was observably lessened following the AAV6–CTRP3 co-treatment (Figure 3A). Moreover, the content of E-cadherin was reduced and that of N-cadherin and α-SMA was boosted in lung tissues by OVA induction. However, these influences were lessened by AAV6–CTRP3 co-treatment (Figure 3B). These outcomes revealed that CTRP3 alleviated OVA-induced airway remodeling in mice.
Figure 3 CTRP3 restricted OVA-induced airway remodeling. (A) The bronchial and alveolar structures were assessed by H&E staining and sirius red staining (×200). (B) Levels of E-cadherin, N-cadherin, and α-SMA were examined by Western blot analysis. Compared to the control group, ***P < 0.001; compared to the OVA+vector group, ^^^P < 0.001. All data were expressed as mean ± SD.
CTRP3 modulated NF-κB and TGFβ1/Smad3 pathways in OVA-induced mice
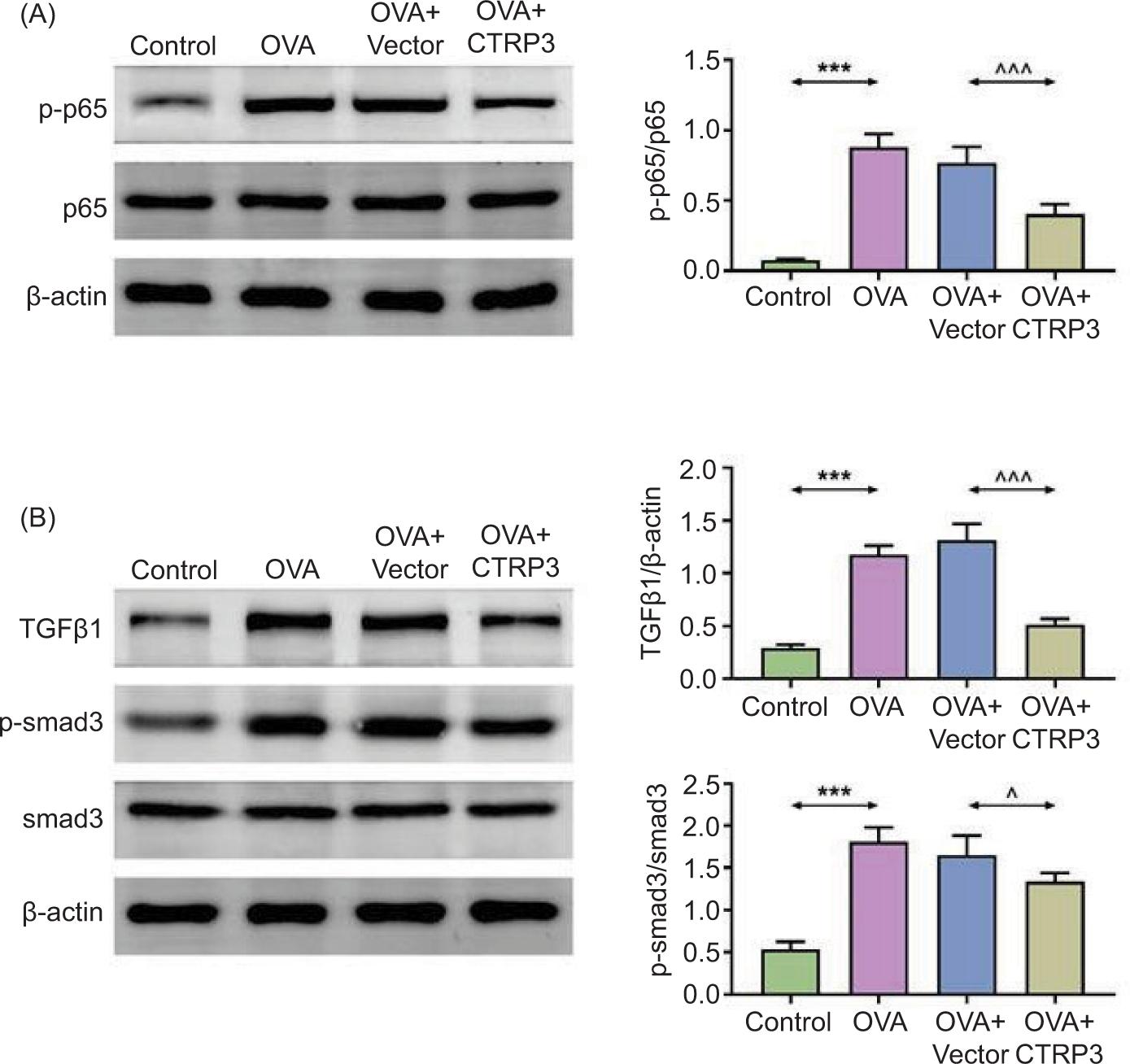
We explored CTRP3-modulated signaling pathway. We found that the contents of p-p65/p65 (Figure 4A), TGFβ1, and p-Smad3/Smad3 (Figure 4B) in lung tissues were increased after OVA induction but decreased by AAV6-CTRP3 co-treatment. Hence, we confirmed that CTRP3 could modulate NF-κB and TGFβ1/Smad3 pathway in OVA-induced mice. The main numerical values are given in Supplementary Table S1.
Figure 4 CTRP3 modulated NF-κB and TGFβ1/Smad3 pathways in OVA-stimulated mice. (A) and (B) The contents of p-p65/p65, TGFβ1, and p-Smad3/Smad3 in lung tissues were examined by Western blot analysis. Compared to the control group, ***P < 0.001; compared to the OVA+vector group, ^P < 0.05, ^^^P < 0.001. All data were expressed as mean ± SD.
Discussion
In this paper, we found that CTRP3 was downregulated in mice of the OVA groups, and AAV6-CTRP3 treatment markedly upregulated the expression of CTRP3. Besides, upregulation of CTRP3 diminished asthmatic airway inflammation by dwindling the number of inflammatory cells and the contents of proinflammatory factors. Moreover, we also found that CTRP3 could markedly lessen AWR and increase lung function in OVA mice. Furthermore, histological analysis found that CTRP3 curbed OVA-induced airway remodeling in mice. Finally, we also investigated the regulatory molecular mechanism of CTRP3 in asthma. CTRP3 modulated NF-κB and TGFβ1/Smad3 pathways in OVA-stimulated mice.
Asthma is a common disease with globally high morbidity, mortality, and economic burden.22 Asthma is a heterogeneous disease associated with many factors, such as infections, tobacco smoke, cold air, allergens, hormones, exercise, obesity, genetic mutations, and eosinophilia.23–26 Meanwhile, asthma is also associated with chronic airway inflammation caused by activation of the immune system.7 Chronic airway inflammation leads to airway edema, excessive secretion of mucus, airway blockage, decrease in lung functioning, and related conditions.27 At present, hormone therapy is often used to treat asthma, but its adverse reactions are severe.28 With the in-depth understanding of the pathophysiological mechanism of asthma, molecular-targeted therapy is gradually applied to treat asthma patients.7 In this research, we scrutinized the functions of CTRP3 in asthma.
CTRP3 has a certain regulatory effect on inflammatory reaction, blood glucose, and blood lipid.14,29,30 Besides, Yu et al. confirmed that CTRP3 could attenuate inflammatory bowel disease (IBD) by modulating NF-κB signaling.31 In addition, Zhang et al. revealed that CTRP3 could ameliorate uric acid-stimulated inflammation in vascular endothelial cells.32 Moreover, Meng et al. proved that the CTRP3 content was obviously abridged in the hippocampus of mice with depression, and CTRP3 curbed inflammatory response in depressive mouse model by modulating NF-κB signaling.33 In addition, CTRP3 participates in the regulation of obesity, metabolic dysfunction, and cardiovascular diseases.34,35 However, no study has essentially investigated the role of CTRP3 in asthma. In this paper, we exposed that CTRP3 was downregulated in OVA groups, and AAV6–CTRP3 treatment markedly upregulated the expression of CTRP3 in OVA-stimulated mice. Upregulation of CTRP3 diminished asthmatic airway inflammation by diminishing the number of inflammatory cells and the contents of proinflammatory factors. These outcomes were consistent with the results of Zhang et al. and Meng et al., which confirmed that CTRP3 has a good anti-inflammatory effect.32,33
Moreover, we found that CTRP3 could markedly lessen AWR and increase lung function in OVA-stimulated mice. Furthermore, the results of H&E staining and sirius red staining found that CTRP3 curbed OVA-induced airway remodeling in mice. Previous studies have exposed that E-cadherin, N-cadherin, and α-SMA were linked with airway remodeling in asthma, and the content of E-cadherin was reduced in asthmatic epithelial cells.36 Pu et al. reported that azithromycin could markedly repress airway remodeling by enhancing the abundance of E-cadherin and decreasing the contents of N-cadherin and α-SMA.37 Herein, we revealed that the content of E-cadherin was restricted. Besides, contents of N-cadherin and α-SMA were increased in lung tissues by OVA induction but lessened by AAV6–CTRP3 co-treatment. These results were comparable with the results of Pu et al.37
NF-κB is a potent inflammatory mediator that enhances cytokine production in a variety of diseases, including asthma.38,39 Phosphorylation of IκB activates the NF-κB p65 subunit, which is responsible for increasing cytokine production and promoting inflammatory cascade reaction.38,40 TGF-β1 is a multipotent cytokine involved in airway inflammation and fibrotic tissue remodeling during asthma pathology. TGF-β1 functions through a number of different signaling pathways, including the Smad pathway. TGF-β1 activates Smad pathway in a nonclassical manner by activating all three known mitogen-activated protein kinase pathways.41 Wu et al. revealed that CTRP3 could exert anti-fibrotic effect by modulating Smad3 activation.14 In this paper, we confirmed that CTRP3 modulated NF-κB and TGFβ1/Smad3 pathways in OVA-stimulated asthmatic mice. We found that CTRP3 could be a genetic target for asthma treatment, with good anti-inflammatory effects for the first time.
The present research has certain drawbacks. OVA are not a “real” asthma allergen in humans, and therefore the OVA model may not accurately reflect the pathophysiology of human asthma. In contrast, Aspergillus fumigatus is a common allergen of human asthma; hence, we would use this allergen for establishing modeling for subsequent confirmation.42 Second, since we only studied the effect of CTRP3 in a mouse model, relevant conclusions need to be further verified in clinical practice.
Conclusion
In summary, our results proved that CTRP3 could alleviate airway inflammation and remodeling in OVA-stimulated asthmatic mice by regulating NF-κB and TGFβ1/Smad3 pathways. Hence, CTRP3 could be a possible therapeutic target for the repression of asthma.
Availability of Data and Materials
All data generated or analyzed during this study are included in this published article.
The datasets used and/or analyzed in the present study are available from corresponding author on reasonable request.
Competing Interests
The authors stated that there were no conflicts of interest to disclose.
Author Contributions
Both authors contributed to the study’s conception and design. Material preparation and experiments were performed by Hai Lin. Data collection and analysis was done by both authors. The first draft of the manuscript was written by Jinrong Yi. Both authors commented on the previous versions of the manuscript. Both authors read and approved the final manuscript.
REFERENCES
1. Padem N, Saltoun C. Classification of asthma. Allergy Asthma Proc. 2019;40(6):385–8. 10.2500/aap.2019.40.4253
2. Poddighe D, Brambilla I, Licari A, Marseglia GL. Omalizumab in the therapy of pediatric asthma. Recent Pat Inflamm Allergy Drug Discov. 2018;12(2):103–9. 10.2174/1872213X12666180430161351
3. Dong L, Wang Y, Zheng T, Pu Y, Ma Y, Qi X, et al. Hypoxic hUCMSC-derived extracellular vesicles attenuate allergic airway inflammation and airway remodeling in chronic asthma mice. Stem Cell Res Ther. 2021;12(1):4. 10.1186/s13287-020-02072-0
4. Bousquet J, Schunemann HJ, Togias A, Bachert C, Erhola M, Hellings PW, et al. Next-generation allergic rhinitis and its impact on asthma (ARIA) guidelines for allergic rhinitis based on grading of recommendations assessment, development and evaluation (GRADE) and real-world evidence. J Allergy Clin Immunol. 2020;145(1):70–80.e3. 10.1046/j.1472-9725.2003.00063.x
5. Cardona V, Ansotegui IJ, Ebisawa M, El-Gamal Y, Fernandez Rivas M, Fineman S, et al. World allergy organization anaphylaxis guidance 2020. World Allergy Organ J. 2020;13(10):10047210.1016/j.waojou.2020.100472
6. Patel SJ, Teach SJ. Asthma. Pediatr Rev. 2019;40(11):549–67. 10.1542/pir.2018-0282
7. Gans MD, Gavrilova T. Understanding the immunology of asthma: Pathophysiology, biomarkers, and treatments for asthma endotypes. Paediatr Respir Rev. 2020;36:118–27. 10.1016/j.prrv.2019.08.002
8. Zhang J, Zhou Y, Gu H, Zhang J, Tang H, Rong Q, et al. LncRNA-AK149641 associated with airway inflammation in an OVA-induced asthma mouse model. J Bioenerg Biomembr. 2020;52(5):355–65. 10.1007/s10863-020-09844-6
9. Jones TL, Neville DM, Chauhan AJ. Diagnosis and treatment of severe asthma: A phenotype-based approach. Clin Med (Lond). 2018;18(Suppl 2):s36–40. 10.7861/clinmedicine.18-2-s36
10. Niessen NM, Gibson PG, Simpson JL, Scott HA, Baines KJ, Fricker M. Airway monocyte modulation relates to tumour necrosis factor dysregulation in neutrophilic asthma. ERJ Open Res. 2021;7(3):131-2021. 10.1183/23120541.00131-2021
11. Li Y, Wright GL, Peterson JM. C1q/TNF-related protein 3 (CTRP3) function and regulation. Compr Physiol. 2017;7(3):863–78. 10.1002/cphy.c160044
12. Wei WY, Ma ZG, Zhang N, Xu SC, Yuan YP, Zeng XF, et al. Overexpression of CTRP3 protects against sepsis-induced myocardial dysfunction in mice. Mol Cell Endocrinol. 2018;476:27–36. 10.1016/j.mce.2018.04.006
13. Lv C, He Y, Wei M, Xu G, Chen C, Xu Z, et al. CTRP3 ameliorates cerulein-induced severe acute pancreatitis in mice via SIRT1/NF-kappaB/p53 axis. Biosci Rep. 2020;40(10):BSR20200092. 10.1042/BSR20200092.
14. Wu D, Lei H, Wang JY, Zhang CL, Feng H, Fu FY, et al. CTRP3 attenuates post-infarct cardiac fibrosis by targeting Smad3 activation and inhibiting myofibroblast differentiation. J Mol Med (Berl). 2015;93(12):1311–25. 10.1007/s00109-015-1309-8
15. National Research Council (US) Committee for the Update of the Guide for the Care and Use of Laboratory Animals. Guide for the Care and Use of Laboratory Animals, 8th ed. Washington, DC: National Academies Press; 2011. Reports funded by National Institutes of Health. PMid: 21595115.
16. Strobel B, Duechs MJ, Schmid R, Stierstorfer BE, Bucher H, Quast K, et al. Modeling pulmonary disease pathways using recombinant adeno-associated virus 6.2. Am J Respir Cell Mol Biol. 2015;53(3):291–302. 10.1165/rcmb.2014-0338MA
17. Liang S, Zhou Z, Zhou Z, Liang J, Lin W, Zhang C, et al. Blockade of CBX4-mediated beta-catenin SUMOylation attenuates airway epithelial barrier dysfunction in asthma. Int Immunopharmacol. 2022;113(Pt A):109333. 10.1016/j.intimp.2022.109333
18. Rahmati M, Shariatzadeh Joneydi M, Koyanagi A, Yang G, Ji B, Won Lee S, et al. Resistance training restores skeletal muscle atrophy and satellite cell content in an animal model of Alzheimer’s disease. Sci Rep. 2023;13(1):2535. 10.1038/s41598-023-29406-1
19. Rahmati M, Taherabadi SJ. The effects of exercise training on Kinesin and GAP-43 expression in skeletal muscle fibers of STZ-induced diabetic rats. Sci Rep. 2021;11(1):9535. 10.1038/s41598-021-89106-6
20. Bostani M, Rahmati M, Mard SA. The effect of endurance training on levels of LINC complex proteins in skeletal muscle fibers of STZ-induced diabetic rats. Sci Rep. 2020;10(1):8738. 10.1038/s41598-020-65793-5
21. Rahmati M, Rashno A. Automated image segmentation method to analyse skeletal muscle cross section in exercise-induced regenerating myofibers. Sci Rep. 2021;11(1):21327. 10.1038/s41598-021-00886-3
22. Rehman A, Amin F, Sadeeqa S. Prevalence of asthma and its management: A review. J Pak Med Assoc. 2018;68(12):1823–7.
23. Miller RL, Grayson MH, Strothman K. Advances in asthma: New understandings of asthma’s natural history, risk factors, underlying mechanisms, and clinical management. J Allergy Clin Immunol. 2021;148(6):1430–41. 10.1016/j.jaci.2021.10.001
24. Wu HK, Chang ES, Cheng CW, Chien WC, Chou HH. Impact of COVID-19 pandemic waves on pediatric emergency department patients presenting with asthma attacks in Taiwan. Signa Vitae. 2022; 18(6):27-32. https://www.signavitae.com/articles/10.22514/sv.2022.045
25. Tanaka K, Arakawa M, Miyake Y. Perinatal smoking exposure and risk of asthma in the first three years of life: A prospective prebirth cohort study. Allergol Immunopathol (Madr). 2020;48(6):530–6. 10.1016/j.aller.2020.03.008
26. Bimstein E, Wilson J, Guelmann M, Primosch RE. The relationship between oral and demographic characteristics of children with asthma. J Clin Pediatr Dent. 2006;31(2):86–9. 10.17796/jcpd.31.2.977776084511l005
27. Cazzola M, Rogliani P, Ora J, Calzetta L, Matera MG. Asthma and comorbidities: Recent advances. Pol Arch Intern Med. 2022;132(4):16250. 10.20452/pamw.16250.
28. Funston W, Higgins B. Improving the management of asthma in adults in primary care. Practitioner. 2014;258(1776):15–9, 2.
29. Hu TY, Li LM, Pan YZ. CTRP3 inhibits high glucose-induced human glomerular mesangial cell dysfunction. J Cell Biochem. 2019;120(4):5729–36. 10.1002/jcb.27859
30. Petersen PS, Wolf RM, Lei X, Peterson JM, Wong GW. Immunomodulatory roles of CTRP3 in endotoxemia and metabolic stress. Physiol Rep. 2016;4(5):e12735. 10.14814/phy2.12735
31. Yu H, Zhang Z, Li G, Feng Y, Xian L, Bakhsh F, et al. Adipokine C1q/tumor necrosis factor-related protein 3 (CTRP3) attenuates intestinal inflammation via Sirtuin 1/NF-kappaB signaling. Cell Mol Gastroenterol Hepatol. 2023;15(4):1000–1015. 10.1101/2022.05.08.491034
32. Zhang J, Lin X, Xu J, Tang F, Tan L. CTRP3 protects against uric acid-induced endothelial injury by inhibiting inflammation and oxidase stress in rats. Exp Biol Med (Maywood). 2022;247(2):174–83. 10.1177/15353702211047183
33. Meng J, Wang DM, Luo LL. CTRP3 acts as a novel regulator in depressive-like behavior associated inflammation and apoptosis by meditating p38 and JNK MAPK signaling. Biomed Pharmacother. 2019;120:109489. 10.1016/j.biopha.2019.109489
34. Guo B, Zhuang T, Xu F, Lin X, Li F, Shan SK, et al. New insights into implications of CTRP3 in obesity, metabolic dysfunction, and cardiovascular diseases: Potential of therapeutic interventions. Front Physiol. 2020;11:570270. 10.3389/fphys.2020.570270
35. Yaribeygi H, Rashidfarrokhi F, Atkin SL, Sahebkar A. C1q/TNF-related protein-3 and glucose homeostasis. Diabetes Metab Syndr. 2019;13(3):1923–7. 10.1016/j.dsx.2019.04.047
36. Turkeli A, Yilmaz O, Karaman M, Kanik ET, Firinci F, Inan S, et al. Anti-VEGF treatment suppresses remodeling factors and restores epithelial barrier function through the E-cadherin/beta-catenin signaling axis in experimental asthma models. Exp Ther Med. 2021;22(1):689. 10.3892/etm.2021.10121
37. Pu Y, Liu Y, Liao S, Miao S, Zhou L, Wan L. Azithromycin ameliorates OVA-induced airway remodeling in Balb/c mice via suppression of epithelial-to-mesenchymal transition. Int Immunopharmacol. 2018;58:87–93. 10.1016/j.intimp.2018.03.016
38. Gai YL, Hao YF, Guo K. PIM2 promotes lung adenocarcinoma cell migration by regulating XIAP/NF-κB pathway. J Men Health. 2021; 17(3):153-159. https://www.jomh.org/articles/10.31083/jomh.2021.052
39. Sheng XJ, Zhou DM, Liu Q, Lou SY, Song QY, Zhou YQ. BRMS1 inhibits expression of NF-kappa B subunit p65, uPA and OPN in ovarian cancer cells. Eur J Gynaecol Oncol. 2014;35(3):236–42.
40. Wang Q, Cui Y, Wu X, Wang J. Evodiamine protects against airway remodelling and inflammation in asthmatic rats by modulating the HMGB1/NF-kappaB/TLR-4 signalling pathway. Pharm Biol. 2021;59(1):192–9. 10.1080/13880209.2020.1871374
41. Huo R, Tian X, Chang Q, Liu D, Wang C, Bai J, et al. Targeted inhibition of beta-catenin alleviates airway inflammation and remodeling in asthma via modulating the profibrotic and anti-inflammatory actions of transforming growth factor-beta(1). Ther Adv Respir Dis. 2021;15:1753466620981858. 10.1177/1753466620981858
42. Poddighe D, Mathias CB, Freyschmidt EJ, Kombe D, Caplan B, Marseglia GL, et al. Basophils are rapidly mobilized following initial aeroallergen encounter in naive mice and provide a priming source of IL-4 in adaptive immune responses. J Biol Regul Homeost Agents. 2014;28(1):91–103.
Supplementary
Table S1. The main numerical values.
| Control | OVA | OVA+vector | OVA+CTRP3 | |
|---|---|---|---|---|
| 1-B | 0.75±0.08 | 0.17±0.02 | 0.21±0.03 | 0.69±0.12 |
| 1-C Total | 2.52±0.1 | 9.8±1.44 | 10.2±1.53 | 3.29±0.47 |
| 1-C Eosinophils | 0.83±0.01 | 5.33±0.54 | 6.18±0.37 | 1.46±0.15 |
| 1-C Neutrophils | 0.82±0.02 | 1.82±0.24 | 1.67±0.22 | 0.98±0.11 |
| 1-C Lymphocytes | 0.51±0.02 | 2.37±0.16 | 2.6±0.24 | 1.36±0.11 |
| 1-D TNF-α | 78.03±2.02 | 422.04±38.75 | 413.98±46.76 | 154.92±34.73 |
| 1-D IL-1β | 33.33±0.88 | 183.62±11.54 | 183.83±25.4 | 79.5±7.94 |
| 2-A PEF | 10.48±1.84 | 6.36±1.07 | 6.52±0.96 | 8.41±0.77 |
| 2-A FEV 0.4/FVC | 0.98±0.11 | 0.57±0.04 | 0.55±0.08 | 0.85±0.06 |
| 2-A Respiratory rate | 118.83±9.41 | 224.17±30.8 | 228.17±17.86 | 155.5±18.9 |
| 2-B 12.5mg/ml | 2.05±0.04 | 6.99±0.77 | 6.4±0.59 | 4.33±0.63 |
| 2-B 25mg/ml | 3±0.08 | 19.41±2.41 | 18.32±1.95 | 7.62±1.15 |
| 2-B 50mg/ml | 6.08±0.18 | 22.47±3.5 | 20.72±1.55 | 12.02±1.11 |
| 3-B E-cadherin | 0.41±0.04 | 0.11±0.01 | 0.11±0.02 | 0.32±0.02 |
| 3-B N-cadherin | 0.05±0 | 0.38±0.04 | 0.34±0.05 | 0.16±0.01 |
| 3-B α-SMA | 0.08±0.01 | 0.39±0.03 | 0.41±0.04 | 0.19±0.03 |
| 4-A p-p65/p65 | 0.29±0.03 | 1.18±0.09 | 1.32±0.15 | 0.52±0.05 |
| 4-B TGFβ1 | 0.08±0.01 | 0.88±0.09 | 0.77±0.11 | 0.41±0.07 |
| 4-B p-smad3/smad3 | 0.54±0.09 | 1.81±0.17 | 1.65±0.23 | 1.34±0.11 |