
Download



Review
Primary immunodeficiency and chronic mucocutaneous candidiasis: pathophysiological, diagnostic, and therapeutic approaches
Natalia Egria, Ana Esteve-Soléb, c, d, Àngela Deyà-Martínezb, c, d, Iñaki Ortiz de Landazuria, Alexandru Vlageaa, c, AP Garcíab, c, d, Celia Cardozoe, Carolina Garcia-Vidale, g, Clara San Bartoloméa, Marta Español-Regoa, L Yiyic, d, e, Xavier Bosch-Amatef, g, J Ferrandof, g, Jordi Yagüea, c, g, Manel Juana, c, g, Laia Alsinab, c, d, g, h*
aImmunology Service, Biomedic Diagnostic Center, Hospital Clínic de Barcelona, Universitat de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
bClinical Immunology and Primary Immunodeficiencies Unit, Pediatric Allergy and Clinical Immunology Department, Hospital Sant Joan de Déu, Barcelona, Spain
cFunctional Unit of Clinical Immunology, Hospital Sant Joan de Déu-Hospital Clínic de Barcelona, Barcelona, Spain
dInstitut de Recerca Sant Joan de Déu, Barcelona, Spain
eInfectious Diseases Service, Hospital Clínic de Barcelona, Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS), Barcelona, Spain
fDermatology Service, Hospital Clínic de Barcelona, Barcelona, Spain
gUniversitat de Barcelona, Barcelona, Spain
hClinical Immunology Working Party of the Sociedad Española de Inmunología Clínica Alergología y Asma Pediátrica (SEICAP), Spain
Abstract
Chronic mucocutaneous candidiasis (CMC) is characterized by a chronic or recurrent non-invasive infection, mainly due to Candida albicans, in skin, nails, and mucous membranes, associated in some cases with autoimmune manifestations. The key immune defect is a disruption of the action of cytokine IL-17, whose most common genetic etiology is STAT1 gene gain-of-function (GOF) mutations. The initial appropriate treatment for fungal infections is with azoles. However, the frequent occurrence of drug resistance is the main limitation. Therefore, identification of the underlying inborn error if immunity in CMC may allow to widen therapeutic options aimed at restoring immunological function. Type I and II Janus kinase-inhibitors have been shown to control CMC in cases associated with STAT1 GOF. In this review, we delve into the pathogenesis of CMC and the underlying immune mechanisms. We describe the reported genetic defects in which CMC is the main manifestation. Diagnostic and therapeutic approaches for these patients are also offered.
Key words: candidiasis, chronic mucocutaneous candidiasis, primary immunodeficiency, IL-17, STAT1 GOF
*Corresponding author: Alsina L, Clinical Immunology and Primary Immunodeficiencies Unit, Pediatric Allergy and Clinical Immunology Department, Hospital Sant Joan de Déu, Barcelona, Spain. Email address: [email protected]
Received 30 October 2020; Accepted 17 November 2020; Available online 2 January 2021
Copyright: Egri N, et al.
License: This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
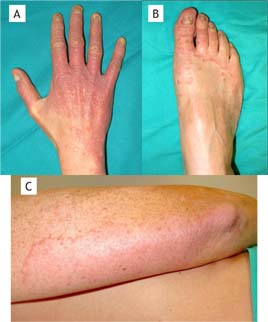
Chronic mucocutaneous candidiasis (CMC) shows a heterogeneous clinical phenotype with common characteristics, including chronic non-invasive infection mainly by Candida albicans, and, less frequently, by C. glabrata, C. tropicalis, and other species such as dermatophyte fungi on the skin, nails, and mucous membranes.1 The first clinical evidence is usually a persistent thrush characterized by one or more white pseudomembranes covering the tongue, soft palate, and buccal mucosa. When these pseudomembranes detach, the reddened, eroded area left exposed may spread and make swallowing difficult. Subsequently, alterations of the coloration, onycholysis, and dystrophy of the nails are seen (Figure 1), mainly affecting the proximal part and the lateral edges or the free edge, depending on whether it is Candida or dermatophytes, respectively. In addition, skin, hair, and mucosa are usually affected.
Figure 1 Chronic mucocutaneous candidiasis in a patient with autoimmune regulator (AIRE) gene mutation. (A) Onychomycosis and chronic cutaneous candidiasis in fingers and back of the right hand. (B) Candidiasic onychopathy and ringworm of the right foot. (C) Ringworm corporis secondary to Trichophyton rubrum infection in the right arm.
Chronic mucocutaneous candidiasis can occur in isolation,2 or associated with bacterial and viral infections, or with autoimmune diseases like endocrinopathies, autoimmune cytopenias, and rheumatoid arthritis, but to a lesser degree. In terms of the evolution of the condition, chronic local inflammation caused by persistent or recurrent Candida infection may predispose to the development of neoplasia (squamous cell carcinoma) mainly affecting the esophagus and the mouth. Rarely, cerebral vasculitis can also occur.3
The first genetic cause of isolated CMC was reported in 2011.4 In fact, isolated CMC can be caused by congenital inborn errors of immunity, thus being included in the primary immunodeficiencies (PID) classification.5
In this review, we explore the pathogenesis of CMC and the underlying immunological mechanisms. We describe the identified PID with CMC as their main manifestation. Additionally, we highlight the recommended diagnostic and therapeutic approaches in patients diagnosed with CMC.
Immunity against Candida
Candida albicans is a polymorphic fungus found in the digestive tract and other mucous surfaces of the body such as the oral cavity and the vagina. It is considered a normal component of the microbiota, but disturbances in the local environment can lead to superficial mucosal infections, mainly in the oral cavity and the vagina. There is a complex and dynamic relationship between Candida albicans and the host whose balance is highly influenced by the effectiveness of the host’s immune response.6
The immune response to Candida infections is made up of several mechanisms, including mucocutaneous protective barriers, innate immunity, and adaptive immunity. Regarding innate immunity, macrophages, neutrophils, and dendritic cells recognize the fungus through pattern recognition receptors (PRRs), such as the Dectin-1 receptor and Toll-like receptors (TLR). TLR4, mannose receptor, and beta-glucan receptor complex Dectin 1/TLR2 recognize mannans, which are the main components of the Candida cell wall.7 Altogether, these receptors participate in the stimulation of cytokine production. Dectin-1 increases the production of TLR2 and TLR4-induced cytokines, such as tumor necrosis factor (TNF). In mouse models, the absence of these receptors leads to greater susceptibility to C. albicans and Pneumocystis jirovecii infections.8 After initial recognition, the adaptive immune response mediated by T helper 17 (Th17) and Th1 T cells is activated, and the continuous retro-activation between innate and adaptive immunity guarantees the complete destruction of the fungus6 (Figure 2).
Figure 2 Immune response against Candida. When C. albicans enters the body, it is recognized by innate cells (phagocytes and dendritic cells) through Dectin-1 receptor and Toll-like receptor 2 (TLR2). Upon recognition, these cells produce signals promoting pre-immune CD4+ T lymphocyte (naive) polarization to effector subgroups: Th17 (mainly, IL-17 and IL-22 production) and Th1 (IFN-γ production). The latter, in turn, retro-activate cells for innate immunity. LC: Langerhans cell; DC: dendritic cell.
IL-17 plays a central role in antifungal defense. It induces the recruitment of neutrophils to the inflammation site and increases the levels of defensins produced by keratinocytes to control the overgrowth of fungi like Candida.9.10 The IL-17 cytokine family consists of six members (IL-17A, IL-17B, IL-17C, IL-17D, IL-17E, IL-17F) and the IL-17 receptor family consists of five members (IL-17RA, IL-17RB, IL-17RC, IL-17RD, IL-17RE). IL-17A and IL-17RA are the original members of the cytokine and receptor families, respectively, and are thus commonly known as IL-17 and IL-17R. IL-17A and IL-17F are produced by Th17 cells. The IL-17 family receptors can form homodimers or heterodimers, and each combination recognizes a type of IL-17 cytokine. For example, the receptor complex formed by IL-17RA/C recognizes IL-17A and IL-17F, while the complex formed by IL-17RA/B recognizes IL-17E. In each case, signaling activates the recruitment of nuclear factor kappa-beta activator 1 (ACT1) as an adapter molecule for subsequent intracellular signaling. The disruption of any element in this complex network can cause poor control of Candida growth. Individuals with an impaired immune response to Candida rarely develop the invasive fungal disease, as other elements of the immune response, such as phagocytosis, remain intact.1
Immune abnormalities underlying CMC
A common feature of CMC patients is in vitro anergy of T lymphocytes when stimulated with Candida antigens. A reduction in the role of IL-17 as the central immune axis has been described in CMC.11 This IL-17 defect may be due to several causes: (1) an overall defect in lymphocyte function (patients with congenital or acquired disorders of T lymphocytes develop CMC within a broader infectious spectrum); (2) a selective defect in Th17 lymphocytes; or (3) a defect in the synthesis or signaling of IL-17. When these defects are congenital, they are included in the so-called PID (Table 1), a group of genetic diseases in which there is a quantitative and/or functional alteration of different mechanisms involved in the immune response. Although most cases of genetically based CMC show an early onset, debuts have also been reported in adult patients.
Table 1 Genetic defects associated with CMC, main related clinical manifestations, and key immunological findings.*
| Disease (OMIM) | Gene | Inheritance pattern | CMC frequency (%) | Associated clinical features | Key immune defect | References |
|---|---|---|---|---|---|---|
| HIES | STAT3 | AD | 83% | Rash or pustules, pneumonia, facial and dental abnormalities, scoliosis, joint hyperextensibility | Decreased IL-17 producing T cells | 12, 13, 14, 15 |
| HIES | ZNF341 | AR | - | Mild dental and facial abnormalities, eczema, bacterial skin and respiratory infections, hyperextensibility of joints, bone fractures | Decreased IL-17 producing T cells | 5, 18 |
| STAT1 GOF | STAT1 | AD | 98% | Isolated CMC (and cutaneous and esophageal carcinomas) or may associate bacterial, viral infections, autoimmune diseases, CNS aneurysms. | Impaired Th17 cell development | 4, 24, 25, 26, 7 |
| APS-1 | AIRE | AR | 70-98% | Hypoparathyroidism, primary adrenal insufficiency | Neutralizing anti- IL-17A, IL-17F, IL-22 antibodies | 9, 8, 10, 4, 11 |
| IL-17F deficiency | IL-17F | AD | 70% | Isolated CMC | Impaired IL-17 signaling | 17 |
| IL-17RA deficiency | IL-17RA | AR | 100% | Recurrent staphylococcal infections | Impaired IL-17 signaling | 7 |
| IL-17RC deficiency | IL-17RC | AR | 100% | Isolated CMC | Impaired IL-17 signaling | 16 |
| ACT1 deficiency | ACT1 | AR | 100% | Superficial staphylococcal infections | 17, 18 |
*In the phenotypic classification of PID in 20205 there are other entities with CMC amongst their manifestations, but the objective of this review is to expose those with CMC as the main feature; therefore, CARD9 deficiency, BCL10 deficiency, RORγT deficiency as well as DOCK8 deficiency and Vici Syndrome have not been included.
APS-1: autoimmune polyglandular syndrome type 1; HIES: hyperimmunoglobulinemia E syndrome; ACT1: nuclear factor kappa-beta activator 1; GOF: gain of function; AD: autosomal dominant; AR: autosomal recessive.
Next, we will focus on PID with CMC as the predominant clinical manifestations, which are those selectively affecting Th17 lymphocyte function and/or IL-17 signaling. These include deficiency of AIRE gene, GOF mutations in signal transducer and activator of transcription 1 (STAT1) gene, mutations in STAT3 and ZNF341 genes, and, less frequently, IL17RA and IL17RC mutations, IL-17F deficiency and kappa-beta nuclear factor activator 1 (ACT1) deficiency have also been described.12 Global T lymphocyte defects are beyond the scope of this review.
Primary immunodeficiencies causing a predominant Th17 lymphocyte defect
Loss-of-function mutation in STAT3 gene
Loss-of-function mutation in STAT3 gene is the genetic cause of 60–70% of cases of autosomal dominant (AD) hyperimmunoglobulinemia E syndrome (HIES), OMIM 147060. STAT3 participates in wound healing, angiogenesis, and immunity processes, with an important role in signal transduction of diverse interleukins such as IL-6, IL-10, IL-17, IL-22, IL-23, and IL-27.
Hyperimmunoglobulinemia E syndrome AD syndrome is a complex PID characterized by elevated serum IgE levels, rash/dermatitis, and recurrent bacterial and/or fungal infections of the skin and respiratory tract.13 This syndrome was first described as Job Syndrome in 1966, due to the presence of recurrent staphylococcal abscesses. Affected patients characteristically have a reduction in the number of Th17 cells owing to the loss-of-function STAT3 gene mutation. This is associated with IL-6, IL-17, and IL-22 defects, and, therefore, subsequent great susceptibility to fungal infections.14
Chronic mucocutaneous candidiasis is observed in up to 83% of HIES AD syndrome-affected patients, with Candida infections in the oral, cutaneous, genital, and nail regions.15
Rashes and pustules are early manifestations during childhood, generally appearing in the first months of life. Another hallmark of the disease is co-infection with Streptococcus pneumoniae, Haemophilus influenza, and Staphylococcus aureus, leading to pneumonia, which can be complicated by bronchiectasis and pneumatocele.
Some characteristic facial features such as rough skin, facial asymmetry, prominent forehead, sunken eyes, broad nasal bridge, and mild prognathism have been reported as emerging with age. Abnormalities in dentition with prolonged retention of the primary teeth are a constant feature. In addition, scoliosis, osteoporosis, and hyperextensibility of the joints may be observed. An increased risk of autoimmune diseases (systemic lupus erythematosus) and lymphoproliferative diseases (non-Hodgkin’s lymphoma) has also been described.16
The clearest diagnostic feature is an increase in serum IgE concentration with normal IgG, IgA, and IgM. However, a normal level of IgE does not always rule out the diagnosis of HIES, depending on the age of the patient, since IgE levels decrease or even normalize during adulthood in this condition.13 Specific antibody responses to encapsulated organisms can be impaired.17 Decreased Th17 cytokine production as well as decreased neutrophil chemotaxis is observed.15
Loss-of-function mutations in the ZNF341 gene
Patients with a recessive form of HIES due to loss-of-function mutations in the ZNF341 gene have been reported.
ZNF341 is a transcription factor that is constitutively expressed in the nucleus, where it is required for inducible STAT3 transcription. ZNF341 deficiency prevents the induction of STAT3 transcription by STAT3 activating cytokines, leading to decreased STAT3 phosphorylation and transcriptional activity. Similar to patients with AD STAT3 mutations, patients with ZNF341 deficiency lack Th17 cells, show Th2 polarization, and have decreased memory B cells.18 Clinically, they have a HIES-like phenotype with mild facial dysmorphia, early-onset eczema, CMC, recurrent bacterial skin and respiratory infections, joint hyperextensibility, bone fractures, and retention of primary teeth.5
STAT1 gene mutation with gain-of-function
STAT1 is involved in both the innate and adaptive immune responses to fungi, viruses, and bacteria. It can be activated by various ligands such as IFN-α, IFN-γ, epidermal growth factor, platelet-derived growth factor, and IL-6. Gain-of-function (GOF) mutations in the STAT1 gene (OMIM 614162) can occur in the spiral binding domain (62%) and in the DNA binding domain (35%),; also, in the transactivation domain, the N-terminal domain, and the Src 2 domain (SH2).3 These mutations lead to an increase in phosphorylation, and in some cases also to altered dephosphorylation with the consequent accumulation of phosphorylated STAT1 molecules in the nucleus. For this reason, there is an increase in STAT1-dependent cytokines, such as IL-6, IFN-α and β, IFN-γ, and IL-27; this last cytokine is an inhibitor of IL-17, which conditions a decrease in Th17 responses and IL-17 and IL-22. There is no clear correlation between the location of the mutation and the severity of clinical symptoms, but the results are contradictory.19,20 However, Leiding et al.21 reported that mutations in the DNA binding domain are associated with a severe clinical course.
Since GOF STAT1 mutations were identified as being responsible for CMC-associated molecular pathogenesis, approximately 350 patients with these mutations have been described. Indeed, it is widely considered to be the most common genetic etiology of CMC, without ethnic or age restrictions. These patients have a decrease in IL-17- producing T lymphocytes, which are essential in the defense against Candida.3
Patients can also develop other fungal infections (Pneumocystis jiroveci, Aspergillus spp, and Cryptococcus neoformans) in the context of CMC. Bacterial infections, mainly those of a sinopulmonary nature caused by Streptococcus pneumoniae, Pseudomonas aeruginosa, Haemophilus influenza, and Staphylococcus aureus, are associated as well. Staphylococcus aureus-related folliculitis is common in these patients, in addition to mycobacterial infections mainly owing to Mycobacterium tuberculosis. Viral infections with Herpes simplex virus (HSV), Varicella Zoster virus (VZV), Cytomegalovirus (CMV), Epstein-Barr virus (EBV), and human Papilloma virus (HPV) have also been described.3,22
Autoimmunity clinical features can develop. Hypo- and hyperthyroidism are the most frequent autoimmune manifestations, followed by type 1 diabetes mellitus (DM), vitiligo, alopecia, psoriasis, lupus erythematosus, pernicious anemia, celiac disease, autoimmune hepatitis, and inflammatory bowel disease. This association with autoimmunity could be explained by the strong IFN-α and IFN-β signaling, as observed in patients treated with recombinant IFN-α and IFN-β and in patients with interferonopathies.3
A small fraction of patients, up to 6% according to the review by Toubiana et al.,3 may develop both cerebral and extracerebral aneurysms. In a group of 274 patients, 17 developed aneurysms (14 cerebral, three extracerebral) and five died from a cerebral hemorrhage, all between the ages of 4 and 34. Cerebral aneurysms are more frequent in patients withautoimmunity manifestations, especially type I DM. Abnormalities in connective tissue, alteration of IL-17 immunity, and cerebral or infectious autoimmune vasculitis are the pathogenic mechanisms that explain the formation of aneurysms. One aspect to keep in mind is that brain aneurysms have not been reported in other CMC-causing mutations, except for STAT3 gene deficiency.7
From the immunological perspective, an increase in STAT1-dependent cytokines is observed. There is an increase in IL-6, IFN α and β, IFN γ, and IL-27. IL-27 is an IL-17 inhibitor, and it decreases IL-17 and IL-22 levels. This STAT1 genetic defect leads to increased Th1 responses and decreased Th17 responses.23 Also, the decrease in memory B lymphocytes and IgG 2 and 4 subtypes, CD4+ and CD19+ cells can be observed, as well as dysregulation of follicular Th lymphocytes and an increase in PD- L1.24
More recently, Tabellini et al.25 demonstrated that NK cells in these patients release fewer IFN-γ in response to IL-15 and proliferate less than controls in response to IL-2 or IL-15. They also have decreased cytolytic activity, thus offering a possible explanation for why patients with GOF STAT1 mutations have difficulty with viral infections, including HSV, VZV, CMV, and HPV. Likewise, Vargas-Hernández et al.26 observed that GOF STAT1 mutation is associated with deterioration in the maturation of NK cells, a decrease in perforin expression, and a decrease in cytolytic function. Interestingly, these effects can be partially reversed in vitro and in vivo with Ruxolitinib treatment. Furthermore, as with other immunodeficiencies involving CMC, patients showed lower production of IL-17 by T cells after ex vivo stimulation with PMA-Ionomycin or Candida.
The prognosis of patients with GOF STAT1 gene mutations is poor, mainly due to recurrent infections, but also to aneurysms, malignancies, and autoimmune manifestations.
Primary immunodeficiencies that cause an IL-17 defect
IL-17F deficiency
Mucosal candidiasis has been reported in several members of a family in Argentina. Affected individuals carried a heterozygous mutation in the IL-17F gene, suggesting an AD pattern of inheritance.4
AIRE gene deficiency
Autoimmune regulator was firstly described in 1994. It is located on the long arm of chromosome 21 and encodes a protein that is expressed in stromal cells of lymphoid tissues, including thymic epithelial cells, and it regulates the transcription of restricted antigens to certain tissues. It is responsible for central immune tolerance.27 AIRE disruption leads to Type I autoimmune polyglandular syndrome (APS-1), also known as ectodermal dystrophy–candidiasis-autoimmune polyendocrinopathy (APECED; OMIM 240300). When there is a defect in AIRE gene, the central tolerance mechanisms fail and self-reactive B cells that generate autoantibodies are released into the periphery. These autoantibodies are directed mostly against endocrine glands but also against IL-17F and IL-17A, blocking their function.28
Type I autoimmune polyglandular syndrome is a rare disease with autosomal recessive inheritance (RA), and with an estimated prevalence of approximately 1:100,000, except in Finland (1:25,000), Sardinia (1:14,000), and among Persian Jews in Israel (1:9000).27 Typically, it is characterized by the development during the childhood of the three following cardinal symptoms: CMC, hypoparathyroidism, and primary adrenal insufficiency (Addison’s disease). Recently, other symptoms have been described that may occasionally debut early such as enamel hypoplasia, enteropathy with chronic diarrhea or constipation, primary ovarian failure, bilateral keratitis, and periodic fever with rash, in addition to autoimmune manifestations such as hepatitis, pneumonitis, nephritis, exocrine pancreatitis, and functional asplenia. These findings should lead the clinician to suspect this entity, mainly in young people.29 Some antibody markers of this disease have been described. Those directed against NALP5 (NACHT leucine-rich-repeat protein 5), expressed in parathyroids, are present only in patients with APS-1 with hypoparathyroidism. Anti-type 1 IFN (IFN-α and IFN-β) antibodies are present in all patients with this entity, although they are not specific since they have been observed in myasthenia gravis with thymoma as well as in patients with mutations in the RAG gene. Diagnosis of APS-1 requires the presence of at least two of the cardinal symptoms (CMC, hypoparathyroidism, and primary adrenal insufficiency), or only one if a brother has been diagnosed with this disease or in the presence of anti-IFN type I antibodies, plus at least, one of the following clinical presentations: CMC, hypoparathyroidism, primary adrenal insufficiency, primary ovarian failure (patients younger than 30 years of age), enamel hypoplasia, periodic rash fever, non-infectious keratitis, and autoimmune hepatitis. All these forms of the presentation should be confirmed with the presence of known pathogenic mutations in AIRE.27,30
Primary immunodeficiencies that cause a defect in IL-17 signaling
Mutations in the following three genes have been identified in primary immunodeficiencies caused by a defect in IL-17 signaling: IL-17RA, IL-17RC, and TRAF3IP2 (encoding ACT1 protein). These genes are related to IL-17RA/C-mediated signaling and induced by IL-17A and IL-17F. Similarly, IL-17RA and TRAF3IP2 mutations also affect IL-17RA/B-mediated signaling induced by IL-17E.
Autosomal recessive mutation in the IL-17A receptor (IL17RA)
This mutation generates a deficiency in IL17RA, which was first reported as the genetic etiology of CMC in 2011. Originally called CANDF5, this condition is now known as immunodeficiency 51.12 Most of the described patients present CMC before 6 months of age and suffer recurrent staphylococcal infections.
IL-17 C receptor deficiency (IL17RC)
Some patients of Turkish descent have been reported with homozygous mutations in IL17RC. They presented an early-onset oral candidiasis.31
Deficiency of activator 1 of nuclear factor kappa-beta (ACT1)
Autosomal recessive mutations have been found in the ACT1 gene. In addition to CMC, these patients show an additional predisposition to superficial staphylococcal infections, including dermatitis and blepharitis.4,23
Diagnostic approach to patients with CMC
A diagnosis of CMC should be considered in patients with fungal infections, mainly by Candida or dermatophytes (infection confirmation by culture), which may be chronic (duration of candidiasis greater than 6 months) or recurrent, and involving nails, skin, and mucous membranes as the main infectious presentation.32 Previously, secondary causes such as prolonged use of antibiotics, systemic steroids (≥10mg/day or less when other immunosuppressive drugs or co-morbidities like DM are present), or inhaled immunosuppressive drugs must be ruled out, along with DM or HIV infection. Once a clinical and microbiological diagnosis of CMC has been confirmed, immunological and genetic studies should be carried out to identify the underlying PID.
Immunological studies traditionally include quantification of lymphocyte populations (T, B, NK cells), proliferation of T lymphocytes against mitogens, and lymphocyte subpopulation analysis of T and B cells, including a Th1/Th2/Th17, together with the determination of total and subclasses of IgG, IgA, IgM, and IgE, and vaccine response against polysaccharide antigens in patients with associated bacterial infections.30 Th17 lymphocyte studies are usually only performed in reference centers.
Following the immunological studies, we also recommend conducting genetic studies for CMC associated mutations. These genetic studies can include all CMC associated described PID genes by using gene panels (especially in forms of CMC with other PID warning signs33 and Table 1), or be directed specifically to STAT1 gene sequencing. The latter could be recommended in cases of isolated CMC and a reduction of Th17 cells (according to age-reference values)34; in these cases, performing a STAT1 phosphorylation/dephosphorylation assay (Figure 3) prior to genetic studies may help evidence the STAT1 gain of function to be confirmed by STAT1 Sanger sequencing. Moreover, STAT1 phosphorylation/dephosphorylation assay should be used to assess the functional impact of an identified STAT1 mutation to confirm the gain of function. A proposed diagnostic algorithm is exposed in Figure 4.
Figure 3 Example of STAT1 phosphorylation and dephosphorylation in a healthy control and a STAT1-GOF patient. STAT1 phosphorylation after IFN-γ stimulation in the presence or absence of staurosporine, a kinase inhibitor, in circulating monocytes is increased in STAT1-GOF patients in comparison with healthy controls, and dephosphorylation is also affected.
Figure 4 CMC diagnostic evaluation. Secondary causes such as HIV, antibiotics, corticosteroids or other immunosuppressive drugs, and diabetes mellitus should be ruled out in patients with chronic or recurrent infections with Candida or dermatophytes. In the absence of these causes, immunological studies and subsequent genetic studies in the search for primary causes of CMC (PID) are indicated. *PID warning signs: Table 1.33 **Th17 lymphocyte studies are usually only performed in reference centers and should be correlated to age-matched reference values.34 ***If STAT1 phosphorylation/dephosphorylation assay is not available (only in reference centers), may proceed to STAT1 sequencing.
Immunological targets in the treatment of CMC
The treatment of CMC is initially based on azoles, with fluconazole as a reference drug. In the case of fluconazole resistance, itrazonazole, voriconazole, or posaconazole can be used depending on the fungigram results. Resistance to at least one azole agent is common in CMC patients.35 It is important to monitor liver enzymes in patients who are treated with these drugs because of the possibility of hepatotoxicity. In refractory cases with diagnosed patients, amphotericin B or echinocandins can be used.36 In acute recurrent infections, preventive or suppressive treatment should be considered to avoid re-infections.
Patients with a STAT1 GOF mutation represent up to 50% of CMC cases, and treatments targeting immunological pathways can be effective. These include:
-
Deacetylated histone inhibitors, which counteract STAT1 hyperphosphorylation, allowing STAT1 molecules to leave the nucleus and restore their normal function. In addition, these inhibitors activate the STAT3 pathway, increasing Th17 responses.37
-
Type I and II Janus kinase inhibitors such as ruxolitinib and baricitinib. Treatment with ruxolitinib has been shown to decrease STAT1 hyperphosphorylation, normalize the differentiation of follicular Th1, and enhance the development, in vitro and in vivo, of Th17 cells.9,10 This can determine the resolution of CMC and autoimmune manifestations in some cases. Also, it partially restores the expression of perforin and the cytotoxic function of NK cells.25,26.
Ruxolitinib is currently approved for the treatment of adults suffering from myelofibrosis and polycythemia vera.38 In a recently published study by Bloomfield et al.,39 ruxolitinib treatment was started in a 12-year-old child carrying a new STAT1 GOF mutation with treatment-resistant oral thrush, recurrent onychomycosis, and bacterial infections. Prior to ruxolitinib treatment, this patient had decreased levels of Th17 cells, Tregs, memory B lymphocytes, and IgG subtypes 2 and 4. Ruxolitinib decreased STAT1 phosphorylation levels and a clinical improvement was observed. This patient discontinued the aforementioned treatment for non-medical reasons, with subsequent recurrence of symptoms and increased STAT1 phosphorylation levels. After reintroducing the drug, CMC improvement and decreased STAT1 phosphorylation were again reported. In addition, an increase in CD4+, CD8+, and IgG2 was observed. There was also a decrease in IFN-γ, but no changes were observed in Th17 cells, Tregs, memory B lymphocytes, or IgG4. A series with ruxolitinib treatment in 11 patients carrying STAT1 GOF mutations along with several patients with STAT3 GOF mutations also showed an improvement in 10 of them. The ruxolitinib failure occurred in a patient with disseminated coccidioidomycosis that was active when ruxolitinib treatment was started.40
Baricitinib treatment improves Candida infections and autoimmune manifestations. In fact, it reduces STAT1 hyperphosphorylation, improves the ability of peripheral blood mononuclear cells (PBMCs) to secrete IL-17A, IL-17F, IL-22, and, in lymphocyte cultures, it decreased the expression of genes that regulate STAT1 such as CXCL9 and CXCL10.41Baricitinib has recently been approved for the treatment of moderate-to-severe active rheumatoid arthritis in adults with an inadequate response or intolerant to disease-modifying drugs.42 The case of a 24-year-old woman with CMC and STAT1 GOF mutation has recently been published. She was affected by recurrent esophageal and oral candidiasis in addition to oral and vaginal ulcers. Treatment with baricitinib was started, achieving improvement in symptoms, without evidence of recurrence during the 8-month follow-up. In functional studies, a decrease in hyperphosphorylation and improvement in PBMC production of IL-17 and IL-22 was shown.15
In summary, the use of type I and II Janus kinase inhibitors such as ruxolitinib and baricitinib may be indicated in patients with a STAT1 GOF mutation with refractory disease (mainly resistant Candida infections and autoimmune disease), in whom transplantation from hematopoietic relatives could have poor results.26,41,43
Despite the encouraging results with type I and II Janus kinase inhibitors, the duration of treatment remains to be elucidated since there are no long-term studies in these patients, and the possibility of rapid recurrence is high when the treatment is suspended. Careful clinical surveillance of these drugs should be carried out due to the possibility of side effects such as serious infections and malignancy.
-
IL-27 and IFN-γ inhibitors to restore normal Th17 function are under investigation.
-
Granulocyte-macrophage colony-stimulating factors (GM-CSF) seem to improve impaired Th17 response. Th17 responses participate in the recruitment of neutrophils to the infection site, but their effects are transitory. Clinical trials are required for further evaluation.37
-
Hematopoietic stem cell transplantation (HSCT) is currently the only curative treatment available that is indicated for all patients with STAT1 GOF mutations with a severe clinical phenotype suffering from serious infectious and non-infectious complications refractory to the treatments described above.44 Published data concerning a cohort of 15 patients who received a non-elective HSCT, with severe symptoms of infections and autoimmunity, are not very encouraging. Only four patients had immune reconstitution with a resolution of the disease, and nine patients died. These data show that HSCT should be performed in stable patients, without active manifestations if possible, to obtain better results. The main HSCT-related problems are complications such as graft loss, graft-versus-host disease, and uncontrolled infections.21
Conclusions
Clinical phenotypes of patients with CMC can be extremely varied, ranging from cases with isolated mucocutaneous fungal involvement to others with viral and bacterial infections in addition to autoimmune manifestations. In all of them, IL-17 plays a pivotal role.
Even though the majority of cases show a childhood onset, many are not diagnosed until adulthood. Therefore, a range of specialists could face this entity, which should be suspected and appropriately treated.
In patients with CMC, ruling out different forms of related PID that may underlie the condition is crucial. Targeted treatment can be established if a causal mutation is identified, and genetic counseling and prevention of long-term complications may be undertaken. The STAT 1 GOF mutation is the most common genetic CMC etiology and for carriers, JAK 1/2 inhibitors are therapeutic options to consider, mainly in cases of azole-refractory Candida infections and autoimmunity.
Acknowledgments
This study was supported by the projects PI15/01094, PFIS0200 (AC16/00025), PI18/00223, and FI19/00208 to LA integrated in the Plan Nacional de I+D+I and co-financed by the ISCIII–Subdirección General de Evaluación y Formento de la Investigación Sanitaria–and the Fondo Europeo de Desarrollo Regional (FEDER), by Pla Estratègic de Recerca i Innovació en Salut (PERIS), Departament de Salut, Generalitat de Catalunya (SLT006/17/00199 to LA), by a 2017 Leonardo Grant for Researchers and Cultural Creators, BBVA Foundation to LA and by a 2017 Beca de Investigación de la Sociedad Española De Inmunología Clínica Alergología y Asma Pediátrica (SEICAP) to LA.
Conflicts of interest
The authors declare that they have no conflicts of interest.
REFERENCES
1. Lilic D. Unravelling fungal immunity through primary immune deficiencies. Curr Opin Microbiol. 2012;15:420–426. 10.1016/j.mib.2012.06.003
2. Puel A, Picard C, Cypowyj S, Lilic D, Abel L, Casanova JL. Inborn errors of mucocutaneus immunity to Candida albicans in humans: a role for IL-17 cytokines. Curr Opin Immunol. 2010;22(4):467–474. 10.1016/j.coi.2010.06.009
3. Toubiana J, Okada S, Hiller J, Oleastro M, Lagos Gomez M, Aldave Becerra JC, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. 2016;127(25):3154–3165. 10.1182/blood-2015-11-679902
4. Puel A, Cypowyj S, Bustamante J, Wright J F, Liu L, Lim HK, et al. Chronic mucocutaneous candidiasis in humans with inborn errors of interleukin-17 immunity*. Science 2011;332: 65–68. 10.1126/science.1200439
5. Bousfiha A, Jeddane L, Picard C, Al-herz W, Ailal F, Chatila T, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol. 2020;40(1):66–81. 10.1007/s10875-020-00758-x
6. Richardson JP, Moyes DL. Adaptive immune responses to Candida albicans infection. Virulence. 2015;6(4):327–337. 10.1080/21505594.2015.1004977
7. Mihai G. Netea, Neil A.R. Gow, Carol A. Munro, Steven Bates, Claire Collins, Gerben Ferwerda, et al. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. 2006;116(6):1642–1650. 10.1172/JCI27114
8. Ferwerda B, Ferwerda G, Platinga TS, Willment J A, van Spriel A B, Venselaar H, et al. UKPMC Funders Group author manuscript human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. 2009;361(18):1760–1767. 10.1056/NEJMoa0901053
9. Mengesha B, Conti H. The role of IL-17 in protection against mucosal Candida infections. J Fungi. 2017;3(4):52. 10.3390/jof3040052
10. Li X, Bechara R, Zhao J, McGeachy MJ, Gaffen SL. IL-17 receptor-based signaling and implications for disease. Nat Immunol. 2019;20(12):1594–1602. 10.1038/s41590-019-0514-y
11. Hernández-Santos N, Gaffen SL. Th17 cells in immunity to Candida albicans. Cell Host Microbe. 2012;11(5):425–435. 10.1016/j.chom.2012.04.008
12. Green L, Dolen WK. Chronic candidiasis in children. Curr Allergy Asthma Rep. 2017;17(5):31. 10.1007/s11882-017-0699-9.
13. Al-Shaikhly T, Ochs HD. Hyper IgE syndromes: clinical and molecular characteristics. Immunol Cell Biol. 2019;97(4):368–379. 10.1111/imcb.12209
14. Schimke LF, Sawalle-Belohradsky J, Roesler J, Wollenberg A, Rack A, Borte M, et al. Diagnostic approach to the hyper-IgE syndromes: immunologic and clinical key findings to differentiate hyper-IgE syndromes from atopic dermatitis. J Allergy Clin Immunol. 2010;126(3):611.e1–617.e1. 10.1016/j.jaci.2010.06.029
15. Davidson L, Netea MG, Kullberg BJ. Patient susceptibility to candidiasis–a potential for adjunctive immunotherapy. J Fungi. 2018;4(1):9. 10.3390/jof4010009
16. Grimbacher B, Holland SM, Puck JM. Hyper-IgE syndromes. Immunol Rev. 2005;203:244–250. 10.1111/j.0105-2896.2005.00228.x
17. Bergerson JRE, Freeman AF. An update on syndromes with a Hyper-IgE phenotype. Immunol Allergy Clin N Am. 2019;39:49–61. 10.1016/j.iac.2018.08.007
18. Béziat V, Li J, Lin J X, Ma C, Li P, Bousfiha A, et al. A recessive form of Hyper IgE syndrome by disruption of ZNF341-dependent STAT3 transcription and activity. Sci Immunol. 2018;3(24):eaat4956. 10.1126/sciimmunol.aat4956.
19. Frans G, Moens L, Schaballie H, Eyck L V, Borgers H, Wuyts M, et al. Gain-of-function mutations in signal transducer and activator of transcription 1 (STAT1): chronic mucocutaneous candidiasis accompanied by enamel defects and delayed dental shedding. J Allergy Clin Immunol. 2014;134(5):1209–1213. 10.1016/j.jaci.2014.05.044
20. Yamazaki Y, Yamada M, Kawai T, Morio T, Onodera M, Ueki M, et al. Two novel gain-of-function mutations of STAT1 responsible for chronic mucocutaneous candidiasis disease: impaired production of IL-17A and IL-22, and the presence of anti-IL-17F autoantibody. J Immunol. 2014;193(10):4880–4887. 10.4049/jimmunol.1401467
21. Leiding JW, Okada S, Hagin D, Abinun M, Shcherbina A, Balashov D, et al. Hematopoietic stem cell transplantation in patients with gain-of-function signal transducer and activator of transcription 1 mutations. J Allergy Clin Immunol. 2017;141(2):704.e5–717.e5. 10.26226/morressier.57bc1754d462b80290b4d4d7
22. Akarcan S, Severcan E, Karaca N, Isik E, Aksu G, Migaud M, et al. Gain-of-function mutations in STAT1: a recently defined cause for chronic mucocutaneous candidiasis disease mimicking combined immunodeficiencies. Case Reports Immunol. 2017;2017:1–6. 10.1155/2017/2846928 10.1155/2017/2676403
23. Boisson B, Wang C, Pedergnana V, Wu L, Cypowyj S, Rybojad M, et al. A biallelic ACT1 mutation selectively abolishes interleukin-17 responses in humans with chronic mucocutaneous candidiasis. Immunity. 2013;39(4):676–686. 10.1016/j.immuni.2013.09.002
24. Jhamnani RD, Rosenzweig SD. An update on gain-of-function mutations in primary immunodeficiency diseases. Curr Opin Allergy Clin Immunol. 2017;17(6):391–397. 10.1097/ACI.0000000000000401
25. Tabellini G, Vairo D, Scomodon O, Tamassia N, Ferraro R S, Patrizi O, et al. Impaired natural killer cell functions in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. 2017;140(2):553.e4–564.e4. 10.1016/j.jaci.2016.10.051
26. Vargas-Hernández A, Mace EM, Zimmerman O, Zerbe C, Freeman A, Rosenzweing S, et al. Ruxolitinib partially reverses functional natural killer cell deficiency in patients with signal transducer and activator of transcription 1 (STAT1) gain-of-function mutations. J Allergy Clin Immunol. 2018;141(6):2142.e5–2155.e5. 10.1016/j.jaci.2017.08.040
27. Husebye ES, Anderson MS, Kämpe O. Autoimmune polyendocrine syndromes. N Engl J Med. 2018;378(2):1132–1141. 10.1056/NEJMra1713301
28. Nwosu I, Oladiran O, Ogbonna-Nwosu C, Anyata A. Autoimmune polyglandular syndrome type 1: a case report and brief review. J Community Hosp Intern Med Perspect. 2019;9(3):252–254. 10.1080/20009666.2019.1616523
29. Zirilli G, Santucci S, Cuzzupé C, Corica D, Pitrolo E, Salzano G. Peculiarities of autoimmune polyglandular syndromes in children and adolescents. Acta Biomed 2017;88(3):271–275. 10.23750/abm.v%vi%i.5898
30. Bonilla FA, Khan DA, Ballas ZK, Chinen J, Frank M M, Hsu J, et al. Practice parameter for the diagnosis and management of primary immunodeficiency. J Allergy Clin Immunol. 2014;136(5):1186.e78–1205.e78. 10.1016/j.jaci.2015.04.049
31. Ling Y, Cypowyj S, Aytekin C, Galicchio M, Camcioglu Y, Nepesov S, et al. Inherited IL-17RC deficiency in patients with chronic mucocutaneous candidiasis. J Exp Med. 2015; 212(5):619–631. 10.1084/jem.20141065
32. Carey B, Lambourne J, Porter S, Hodgson T. Chronic mucocutaneous candidiasis due to gain-of-function mutation in STAT1. Oral Dis. 2019;25(3):684–692. 10.1111/odi.12881
33. García-García A, Gereda-Martínez D, Deyà-Martínez A, Alsina L. The new scenario of primary immunodeficiencies and the role of the clinical immunologist in the specialised clinic]. An Pediatr (Barc). 2020 Feb;92(2):117-118. doi: 10.1016/j.anpedi.2019.09.006. Epub 2019 Nov 7.
34. Garcia-Prat M, Alvarez-Sierra D, Aguiló-Cucurull A, Salgado-Perandrés S, Briongos-Sebastian S, Franco-Jarava C, et al. Extended immunophenotyping reference values in a healthy pediatric extended immunophenotyping reference values in a healthy pediatric population. Cytom Part B Clin Cytom. 2019;96B:223–233. 10.1002/cyto.b.21728
35. Gavino C, Cotter A, Lichtenstein D, Lejtenyi D, Fortin C, Legault C, et al. CARD9 deficiency and spontaneous central nervous system candidiasis: complete clinical remission with GM-CSF therapy. Clin Infect Dis. 2014;59(1):81–84. 10.1093/cid/ciu215
36. Rautemaa R, Richardson M, Pfaller MA, Perheentupa J, Saxén H. Activity of amphotericin B, anidulafungin, caspofungin, micafungin, posaconazole, and voriconazole against Candida albicans with decreased susceptibility to fluconazole from APECED patients on long-term azole treatment of chronic mucocutaneous candidiasis. Diagn Microbiol Infect Dis. 2008;62(2):182–185. 10.1016/j.diagmicrobio.2008.05.007
37. van de Veerdonk FL, Netea MG. Treatment options for chronic mucocutaneous candidiasis. J Infect. 2016;72:S56–S60. 10.1016/j.jinf.2016.04.023
38. The European Medicines Agency – EMA. Jakavi: Ficha tecnica o resumen de las caracteristicas del producto 1. Agencia Eur Medicam. 2006. https://www.ema.europa.eu/en/documents/product-information/jakavi-epar-product-information_es
39. Bloomfield M, Kanderová V, Paračková Z, Vrabcová P, Svaton M, Fronková E, et al. Utility of ruxolitinib in a child with chronic mucocutaneous candidiasis caused by a novel STAT1 gain-of-function mutation. J Clin Immunol. 2018:38(5);589–601. 10.1007/s10875-018-0519-6
40. Forbes LR, Vogel TP, Cooper MA, Castro-Wagner J, Shussler E, Weinacht K, et al. Jakinibs for the treatment of immune dysregulation in patients with gain-of-function signal transducer and activator of transcription 1 (STAT1) or STAT3 mutations. J Allergy Clin Immunol. 2018;142(5):1665–1669. 10.1016/j.jaci.2018.07.020
41. Meesilpavikkai K, Dik WA, Schrijver B, Nagtzaam N, Posthumus-van Sluijs S, van Hagen M, et al. Baricitinib treatment in a patient with a gain-of-function mutation in signal transducer and activator of transcription 1 (STAT1). J Allergy Clin Immunol. 2018;142(1):328.e2–330.e2. 10.1016/j.jaci.2018.02.045
42. The European Medicines Agency – EMA. Olumiant: Ficha tecnica o resumen de las caracteristicas del producto 1. Agencia Eur Medicam. 2006 https://www.ema.europa.eu/en/documents/product-information/olumiant-epar-product-information_es.
43. Weinacht KG, Charbonnier LM, Alroqi F, Plant A, Qiao Q, Wu H, et al. Ruxolitinib reverses dysregulated T helper cell responses and controls autoimmunity caused by a novel signal transducer and activator of transcription 1 (STAT1) gain-of-function mutation. J Allergy Clin Immunol. 2017;139(5):1629.e2–1640.e2. 10.1016/j.jaci.2016.11.022
44. Aldave JC, Cachay E, Núñez L, Chunga A, Murillo S, Cypowyj S, et al. A 1-year-old girl with a gain-of-function STAT1 mutation treated with hematopoietic stem cell transplantation. J Clin Immunol. 2013;33(8):1273–1275. 10.1007/s10875-013-9947-5