
Download






ORIGINAL ARTICLE
Exploring genetic linkage between rheumatoid arthritis and systemic lupus erythematosus through biological networks and prioritizing omega-3 fatty acids as a potent therapeutic
Muhammad Naveeda*, Syed Murtaza Alia, Tariq Azizb*, Syeda Izma Makhdooma, Sana Rehman Cheemaa, Mariam Abdulaziz Alkhateebc, Maher S. Alwethaynanid, Seham O. Alsulamie, Hanan Abdulrahman Saginif, Omniah A. Mansourif
aDepartment of Biotechnology, Faculty of Science and Technology, University of Central Punjab, Lahore, Pakistan
bLaboratory of Animal Health, Hygiene and Food Quality, University of Ioannina Arta Greece
cDepartment of Biology, College of Science, Princess Nourah bint Abdulrahman University, P.O. Box 84428, Riyadh 11671, Saudi Arabia
dDepartment of Clinical Laboratory Sciences, College of Applied Medical Sciences, Shaqra University, Alquwayiyah, Riyadh, Saudi Arabia
eDepartment of Biochemistry, Faculty of Sciences, University of Tabuk, Saudi Arabia
fDepartment of Biological Sciences, College of Sciences, University of Jeddah, Saudi Arabia
Abstract
Rheumatoid arthritis (RA) and systemic lupus erythematosus (SLE) are chronic autoimmune diseases characterized by persistent inflammation and progressive tissue damage, posing significant challenges to effective treatment. To gain deeper insights into their shared molecular mechanisms, we performed an integrative bioinformatics investigation aimed at uncovering common pathways and therapeutic targets. Using a cutoff of p-value < 0.05 and Log2 FC > 1, differential gene expression analysis identified 1178 DEGs in RA and 7783 DEGs in SLE, with 358 genes common to both diseases. Construction of a protein–protein interaction network revealed several hub genes, including PDE4A, H1-10, H4C6, and PIP, which were highly interconnected and clustered into functional modules. Molecular docking analysis demonstrated that alpha linolenic acid (ALA) exhibited strong binding affinity toward these key proteins, with binding energies ranging from –8.3 to –9.4 kcal/mol. Toxicity profiling further suggested that ALA possesses a favorable safety profile, showing minimal risks of hepatotoxicity, neurotoxicity, and related adverse outcomes. Functional enrichment pointed to the involvement of common signaling cascades, particularly the cAMP and TGF-β pathways, as potential therapeutic avenues. Collectively, our findings highlight ALA as a promising therapeutic candidate capable of modulating shared molecular drivers in RA and SLE. Further in vitro and in vivo validation is essential to confirm its mechanistic effects and therapeutic potential for clinical translation.
Key words: rheumatoid arthritis (RA), systemic lupus erythematosus (SLE), differentially expressed genes (DEGs), alpha linolenic acid (ALA), cAMP and TGF-β signaling pathways
*Corresponding authors: Muhammad Naveed, Department of Biotechnology, Faculty of Science and Technology, University of Central Punjab, Lahore 54590, Pakistan. Email address: [email protected]; Tariq Aziz, Laboratory of Animal Health, Hygiene and Food Quality, University of Ioannina Arta Greece. Email address: [email protected]
Received 30 July 2025; Accepted 18 September 2025; Available online 1 November 2025
Copyright: Naveed M, et al.
This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Rheumatoid arthritis (RA) is an autoimmune disease that causes joint inflammation, pain, and damage. It is also known as a systematic inflammatory disease that may be chronic. In this condition, the immune system harms its own body’s healthy tissue, which leads to synovium (inflammation in the lining of joints). Other than joint damage, RA may harm extra-articular organs like lungs, heart, kidneys, digestive system, skin, eye, and the nervous system. The prevalence of RA in the general population is 0.5 to 2%, and its symptoms are joint pain, especially in smaller joints like hands and feet; swelling; and stiffness.1,2
It has been proved that having a family record of RA raises a person’s probability of developing the disease, and the inheritance of RA has been expected to be around 60%. Initial studies and linkage analyses to recognize disease-associated genes reliably highlighted one gene family in specific, the HLA DRB1 allele, which originates within the human MHC on chromosome 6. The HLA section has class I and class II genes, the end of which (HLA-DR, DQ, and DP) produces the alpha and beta chains of the parallel MHC class II molecule. This molecule is found on antigen-presenting cells (APCs) and is accountable for the presentation of extracellular pathogens to T cells, causing an immune response.
Systemic lupus erythematous (SLE) is a heterogeneous autoimmune disease affecting different body organs and systems. The term lupus means wolf in Latin. It is named as such because of facial lesions observed in the disease process that are reminiscent of a wolf’s bite. Like RA disease, SLE is also a chronic disease that harms one’s own body’s healthy tissue of multiple organs which leads to inflammation and damage. The prevalence of SLE disease in the general population is around 5 to 50 cases per 100,000 individuals. Common symptoms of SLE are joint pain, skin rashes (like the butterfly-shaped rash on the face), fatigue, fever, and organ difficulties including the kidney, heart, lungs, or brain. While the precise cause of SLE remains unclear, it’s believed to be a combination of genetic, environmental, and hormonal factors. Many genetic links are unidentified to treat SLE disease. These links may open the potential for complete treatment of SLE.3,4
SLE is a complicated autoimmune disease described by immune system dysfunction, apparent by the body’s attack on its own tissues. The genetic component in SLE is considerable, assessed at around 66% inheritance in studies, still it doesn’t obey a clear Mendelian inheritance pattern. Although the major histocompatibility complex (MHC), specifically the human lymphocyte antigens (HLA), on chromo-some 6, was the main identified genetic link to SLE, studies now imply a multifactorial genetic background. Large-scale studies containing several SLE patients and families have permitted genome-wide examinations, revealing over 100 genetic loci related with SLE vulnerability around various populations. These researches highlight the connection of several genes rather than a single gene. Remarkably, genes involved in B-cell receptor signaling, as well regulatory factor 5 (IRF5)-TNPO3 region, have shown consistent associations with SLE risk across different populations.
RA and SLE are complex autoimmune diseases characterized by chronic inflammation and tissue damage.5 Despite extensive research, effective treatments for these conditions remain a challenge. Omega-3 fatty acids, including alpha linolenic acid (ALA), eicosapentaenoic acid (EPA), and docosahexaenoic acid (DHA), have gained attention for their potential therapeutic benefits.6 These fatty acids possess anti-inflammatory properties that can help alleviate symptoms and modulate immune responses associated with RA and SLE.7 By reducing the production of pro-inflammatory cytokines and modulating immune cell function, Omega-3 fatty acids may mitigate joint inflammation, pain, and systemic manifestations of these diseases.8 Furthermore, Omega-3 fatty acids have been shown to interact with key genetic pathways implicated in RA and SLE, such as those involving HLA-DRB1 alleles and IRF5, potentially influencing disease susceptibility and progression.9 Clinical evidence supports the efficacy of Omega-3 fatty acid supplementation in improving clinical outcomes and enhancing the quality of life for patients with RA and SLE.10 Overall, Omega-3 fatty acids represent a promising adjunctive therapy for managing RA and SLE, offering a safe and well-tolerated option to complement existing treatment strategies.11
The exploration of differentially expressed genes (DEGs) in RA and SLE demands a systematic approach, as both conditions share overlapping clinical manifestations, yet their genetic linkage and common molecular drivers remain poorly understood. Despite the availability of transcriptomic data, limited studies have integrated large-scale bioinformatics to uncover shared gene networks that could explain the mechanistic convergence between RA and SLE. In this study, microarray data relevant to RA and SLE were retrieved from NCBI-GEO using disease-specific keywords. Differential gene expression analysis was performed via NCBI-GEO2R, applying a stringent cutoff of p-value < 0.05 and Log2 FC > 1. Venn diagram analysis using Intervene, a web tool, identified the common DEGs across the two diseases. To further elucidate functional associations, biological interaction networks were constructed in Cytoscape with the STRING query app (confidence level 0.7), while CytoNCA was employed to prioritize central hub proteins. Functional enrichment was conducted using the DAVID database to highlight key pathways involved. Addressing the knowledge gap in therapeutic interventions, molecular docking using HDock was employed to evaluate potential interactions of prioritized proteins with Omega-3 fatty acids, followed by molecular dynamics simulations through MD Web to validate binding stability. This integrative framework aims to bridge the gap in understanding genetic linkage between RA and SLE while prioritizing Omega-3 fatty acids as a promising therapeutic strategy targeting common disease mechanisms.
Methodology
Data acquisition and identification of DEGs
The NCBI-GEO is a website where the microarray data of diseases were retrieved by adding keywords such as RA disease and SLE disorder. From multiple datasets, filters such as Homo sapiens, expression by array, and a number of samples were used to select the data and validate the analysis. The NCBI-GEO2R is a website that allows users to analyze DEGs in a GEO dataset. The datasets of both diseases were analyzed to identify their DEGs. The samples in the datasets were divided into two groups: control and diseased. The Benjamini & Hochberg (False discovery rate) method was applied. The significance of cutoff p-value was set smaller than 0.05, and the Log2 FC threshold value was greater than 1.
Analyzing the common DEGs in both diseases
To find the common DEGs in both diseases, Intervene was utilized. The upregulated and downregulated genes of both datasets were uploaded. A Venn diagram was generated showing the common upregulated and downregulated DEGs.
Protein–protein interaction (PPI) network analysis
Cytoscape is a tool that is used to find the interaction network of different genes and proteins by making the nodes and edges of genes. In the Cytoscape, the String query app was selected and the common DEGs were uploaded. The parameters were set in such a way that the confidence level selected to 0.7 and 50 were used as the maximum number of interactions.
Identification of modules and hubs and their functional enrichment analysis
CytoNCA is another app in Cytoscape that is used to identify important proteins and nodes by performing centrality analysis. Two centrality methods which are betweenness and degree were used by CytoNCA to calculate the node scores. For this centrality analysis, the betweenness and degree without weight were selected and analyzed.
To find the functional enrichment analysis of genes, the DAVID database was utilized. The major KEGG pathway and gene ontology (GO) analysis targeting the three aspects, namely, molecular functions (MF), cellular components (CC), and biological processes (BP), were analyzed.
In addition, cytoscape’s molecular complex detection (MCODE) app was used to examine the functional modules. MCODE was used to find the compact sites of the PPI network by performing graph-theoretic clustering. To find modules, the parameters were modified as: degree cutoff = 2, k-score = 2, max depth = 100, and node value cutoff = 0.2. After getting the modules, the module having a maximum number of nodes was selected to analyze functional enrichment. To further analyze the functional annotation, Module 1 was selected since it had a good number of nodes that could pass the threshold of cutoff value to analyze the GO and KEGG pathway.
Retrieval of common interacting gene protein data bank (PDB)
The common interacting genes implicated in both RA and SLE, identified through Cytoscape analysis, include PDE4A, H1-10, H4C6, and PIP. The structural data for these genes were retrieved from the PDB with the following accession IDs: PDE4A (7K6O), H1-10 (3IJ1), H4C6 (3I8V), and PIP (3ES6).
Retrieval of Omega 3-fatty acids (PubChem)
The role of Omega-3 fatty acids in the treatment of RA and SLE was investigated, focusing on three specific Omega-3 fatty acids: ALA, EPA, and DHA. These fatty acids, with PubChem IDs 5280934, 446284, and 445580, respectively, were retrieved from PubChem for their potential to interact with common genes implicated in both RA and SLE. The genes identified through Cytoscape analysis, which play a role in these autoimmune diseases, include PDE4A, H1-10, H4C6, and PIP.
Molecular docking
Molecular docking studies were conducted using HDock to investigate the interactions between the receptor proteins PDE4A (PDB ID: 7K6O), H1-10 (PDB ID: 3IJ1), H4C6 (PDB ID: 3I8V), and PIP (PDB ID: 3ES6), and the Omega-3 fatty acids ALA (PubChem ID: 5280934), EPA (PubChem ID: 446284), and DHA (PubChem ID: 445580). HDock employs a hybrid algorithm integrating template-based modeling and ab initio free docking to predict ligand–protein binding. Prior to docking, protein structures were energy-minimized, water molecules were removed, and polar hydrogens were added, while ligands were retrieved in 3D SDF format from PubChem and optimized to their lowest energy conformations. Docking was performed using default grid parameters, centered on the predicted active sites of each protein as identified by the software. Binding affinity scores and interaction profiles were used to rank docking poses, with the lowest energy conformations considered as the most reliable complexes. To validate docking results, interaction analyses including hydrogen bonding, hydrophobic interactions, and van der Waals contacts were performed, providing additional confidence in the predicted binding orientations. This rigorous approach enabled the identification of ALA as exhibiting consistently favorable binding affinities across multiple protein targets.
Molecular dynamic simulations
The best docked complex, as determined from the HDock results, underwent molecular dynamics (MD) simulations using the MD Web tool to evaluate the stability and dynamic behavior of the ligand–receptor interactions in a physiological environment. MD Web provides a user-friendly platform to perform MD simulations, incorporating standard CHARMM force field. The system was solvated in a water box with appropriate ions to neutralize the charge. The simulation was run for an extended period, typically in the range of 100 nanoseconds with a temperature of 310K, to ensure comprehensive sampling of the conformational space. The trajectories were analyzed for key parameters including root-mean-square fluctuation (RMSF) and contact Map to confirm the stability of the docked complex.
Toxicity analysis
The toxicity profile of the best interacting ligand was evaluated using ProTox-II, a virtual lab for the prediction of small molecule toxicity. ProTox-II utilizes a combination of molecular similarity, fragment propensities, and machine learning–based models to predict various toxicity endpoints including acute toxicity, hepatotoxicity, cytotoxicity, carcinogenicity, mutagenicity, and immunotoxicity. The ligand structure was submitted to ProTox-II, which provided a toxicity class, LD50 value, and predictions for specific organ toxicities. This analysis helped in understanding the safety profile of the ligand, highlighting any potential risks associated with its therapeutic use.
Results
Analyzation and identification of DEGs
Two datasets GSE10500 and GSE52471 having gene expression profiling by array were selected for the identification of DEGs for RA and SLE, respectively. GSE10500 dataset was developed on platform GPL8300 (Homo sapiens). The number of samples used for the analysis of RA included five diseased persons and three healthy controls. Patients and control samples were enlisted from the analysis of synovial fluid macrophages from five patients with RA.
The GSE5247 dataset was developed using expression profiling by the array. The platform used for this development was GPL571 having HG-U133A_2 array of Affymetrix Human Genome. For the gene expression analysis, 25 number of SLE samples and 13 healthy tissue samples were taken.
For examination of DEGs in each dataset, the log2 FC (fold change) > 1 and adj. p-value < 0.05 were applied. The gene expression analysis showed that there were overall 1178 DEGs with 549 upregulated and 611 downregulated genes for RA (Figure 1A). However, expression analysis in SLE showed that there were 7783 DEGs with overall 743 upregulated and 202 downregulated genes (Figure 1B). Among these DEGs, there were a total of 358 genes (182 upregulated and 176 downregulated) common in both diseases (Figure 1C and D).
Figure 1 The volcano plots of DEGs (A depicts DEGs in RA, and B depicts DEGs in SLE). Red dots indicate upregulated genes, blue dots indicate downregulated genes, and black dots indicate genes that remained unchanged. The Venn diagram in C depicts the common upregulated genes, and D depicts the downregulated genes in both diseases.
PPI analysis
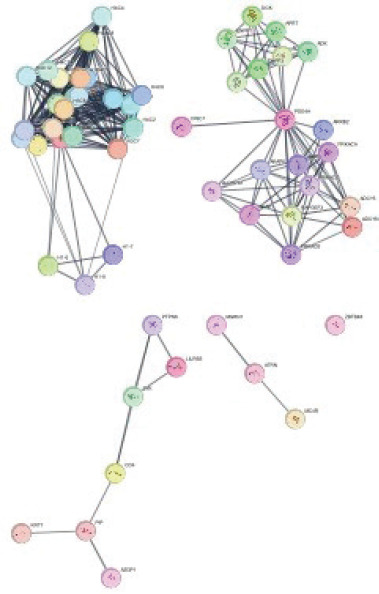
A PPI network was constructed using STRING with the four DEGs common to RA and SLE. The confidence level was set to 0.7, and the maximum number of interactors was limited to 50, yielding a network of 56 nodes and 328 edges (Figure 2). To identify hub genes, network topology was assessed based on degree centrality and betweenness centrality. Among the nodes, PDE4A ranked first with the highest betweenness score (174.13) and a degree score of 19, indicating its strong regulatory role in connecting different clusters. H1-10 followed with a betweenness score of 65 and the highest degree value (24), reflecting its central role in maintaining network connectivity. H4C6 showed a betweenness score of 19 with a degree score of 22, while PIP demonstrated a betweenness score of 18 with a degree score of 3. These results suggest that PDE4A and H1-10 function as major bottlenecks and highly connected hubs, whereas H4C6 and PIP, despite lower betweenness, are embedded within relevant subclusters. Collectively, these metrics highlight PDE4A, H1-10, H4C6, and PIP as key hub genes potentially driving shared molecular mechanisms in RA and SLE.
Figure 2 Protein–protein interaction network analysis of DEGs.
GO analysis
Four genes obtained from the PPI network were subjected to GO and KEGG pathway enrichment using WebGestalt, applying a cutoff of at least four genes. KEGG pathway analysis revealed five significantly enriched pathways (Figure 3). The morphine addiction pathway showed the highest enrichment (enrichment ratio ≈ 38, p < 0.01), followed by parathyroid hormone synthesis, secretion, and action (enrichment ratio ≈ 33, p < 0.01). Osteoclast differentiation was also significantly enriched (enrichment ratio ≈ 26, p < 0.05), along with purine metabolism (enrichment ratio ≈ 20, p < 0.05) and the cAMP signaling pathway (enrichment ratio ≈ 18, p < 0.05). These findings suggest that the shared genes from the PPI network are strongly associated with signaling pathways relevant to immune regulation, bone remodeling, and cyclic nucleotide metabolism.
Figure 3 (A) Major KEGG pathways involved in the top interacted genes. The major gene ontology included are: (B) cellular components, (C) molecular functions, and (D) biological processes.
In terms of GO, enrichment was observed in multiple categories. Within CC, enriched terms included nucleo-some, DNA packaging complex, ruffle membrane, chromo-some telomeric region, and protein–DNA complex. For MF, the top enriched categories were double-stranded telomeric DNA binding, IgG binding, 3’,5’-cyclic-AMP phosphodiesterase activity, immunoglobulin binding, cAMP binding, and aspartic-type endopeptidase activity. Regarding BP, enriched terms included cAMP catabolic process, cyclic nucleotide catabolic process, nucleosome positioning, negative regulation of chromatin silencing, regulation of protein kinase A signaling, negative regulation of T cell apoptotic process, and cAMP metabolic process (Figure 3).
Modules and hubs
Calculation of betweenness and degree centrality was done to examine the arrangement of assembled PPIs. The genes showing high betweenness and centrality values were recognized by CytoNCA, resulting in the formation of hubs (Table 1).
Table 1 Protein–protein interaction network ranking is based on betweenness and degree centrality scores.
| Rank | Gene | Betweenness Score |
Degree Score |
|---|---|---|---|
| 1. | PDE4A | 174.13 | 19 |
| 2. | H1-10 | 65 | 24 |
| 3. | H4C6 | 19 | 22 |
| 4. | PIP | 18 | 3 |
MCODE plug-in was used to identify the modules of the PPI network, and only modules with scores above 4 are selected. Two modules were selected for results: module 1 contained 22 nodes and 231 edges; module 2 contained 17 nodes and 71 edges, which had MCODE scores of 22.000 and 8.875, respectively (Figure 4). The functional enrichment analysis of module 2 showed that six genes were mainly related to response to stimulus in biological processes, membrane component in a cellular component, and protein binding in molecule function (Figure 5A). The major KEGG pathway includes various types of N-glycan biosyn-thesis (Figure 5B).
Figure 4 MCODE modules with interactions. Module 1 with a 22.000 score, 22 nodes, and 231 edges. Module 2 with a 8.875 score, 17 nodes, and 71 edges.
Figure 5 Figure 5 (A) Gene ontology of module 2 genes. The red color indicates the biological processes, blue indicates cellular components, and green indicates the molecular functions of genes. (B) Major KEGG pathways involved include the B-cell receptor signaling, T-cell receptor signaling, NF-κB, cytokine–cytokine receptor interaction, and Toll-like receptor signaling pathways, all of which are critical in autoimmune disease progression.
Molecular docking
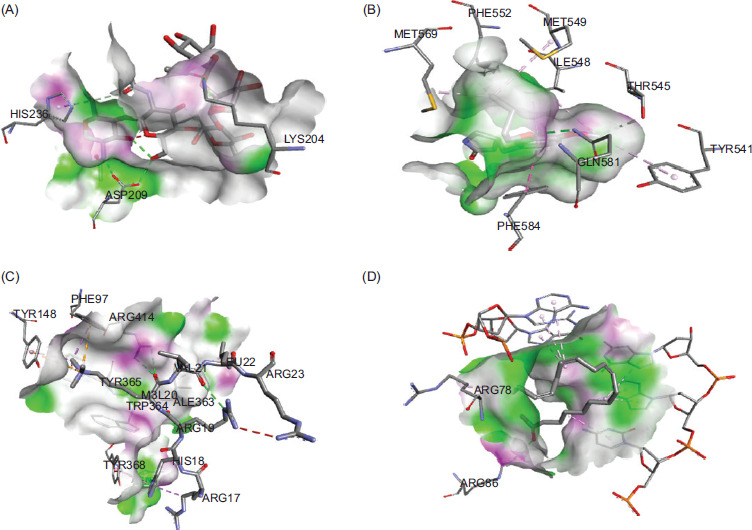
The docking study revealed that ALA demonstrated the best interaction energy with PDE4A, H1-10, H4C6, and PIP, with binding energies of −8.3 kcal/mol (Figure 6A), −9.4 kcal/mol (Figure 6B), −9.1 kcal/mol (Figure 6C), and −8.7 kcal/mol (Figure, 6D) respectively. In comparison, EPA and DHA exhibited binding energies of −7 kcal/mol with all genes. These results suggest that ALA is the most effective Omega-3 fatty acid for potential use as a therapeutic supplement in the treatment of RA and SLE.
Figure 6 Molecular docking interaction energies of ALA with (A) PDE4A, (B) H1-10, (C) H4C6, and (D) PIP, showing superior binding affinities compared to Eicosapentaenoic Acid (EPA) and Docosahexaenoic Acid (DHA).
Molecular dynamic simulations
The RMSF and contact map plots from the MD simulations of ALA with the common interacting genes associated with RA and SLE are presented in Figure 7 (A–D) and Figure 8 (A–D), respectively. These plots demonstrate that the interactions and bonds formed between ALA and the proteins PDE4A, H1-10, H4C6, and PIP are stable throughout the simulation period.
Figure 7 RMSF plots from MD simulations of alpha linolenic acid (ALA) with (A) PDE4A, (B) H1-10, (C) H4C6, and (D) PIP, indicating the stability of the protein–ligand interactions.
Figure 8 Contact map plots from MD simulations of alpha linolenic acid (ALA) with (A) PDE4A, (B) H1-10, (C) H4C6, and (D) PIP, showing stable bond formation between ALA and the proteins.
Toxicity Analysis
The toxicity analysis of ALA using ProTox-II revealed that it is inactive for hepatotoxicity with a probability of 0.54, neurotoxicity with a probability of 0.92, nephrotoxicity with a probability of 0.50, respiratory toxicity with a probability of 0.72, and cardiotoxicity with a probability of 0.98. For toxicity endpoints, ALA is predicted to be inactive for carcinogenicity (0.63), immunotoxicity (0.99), mutagenicity (0.95), cytotoxicity (0.71), and clinical toxicity (0.61), while it is active for the blood–brain barrier (BBB) with a probability of 0.88 and ecotoxicity with a probability of 0.58. It is also inactive for nutritional toxicity with a probability of 0.82.
Regarding the Tox21-Nuclear receptor signaling pathways, ALA is inactive for the aryl hydrocarbon receptor (AhR), androgen receptor (AR), AR ligand-binding domain (AR-LBD), aromatase, estrogen receptor (ER) alpha, ER-ligand binding domain (ER-LBD), and active for peroxisome proliferator-activated receptor gamma (PPAR-Gamma). It is inactive for the nuclear factor (erythroid-derived 2)-like 2/antioxidant responsive element (nrf2/ ARE), heat shock factor response element (HSE), mitochondrial membrane potential (MMP), phosphoprotein (tumor suppressor) p53, and ATPase family AAA domain-containing protein 5 (ATAD5) in the Tox21-Stress response pathways.
In terms of molecular initiating events, ALA is inactive for thyroid hormone receptor alpha (THRα), thyroid hormone receptor beta (THRβ), transthyretin (TTR), ryanodine receptor (RYR), GABA receptor (GABAR), gluta-mate N-methyl-D-aspartate receptor (NMDAR), alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate receptor (AMPAR), kainate receptor (KAR), acetylcholinesterase (AChE), constitutive androstane receptor (CAR), pregnane X receptor (PXR), NADH-quinone oxidoreductase (NADHOX), voltage-gated sodium channel (VGSC), and Na+/I- symporter (NIS). For metabolism-related endpoints, ALA is inactive for cytochrome CYP1A2, CYP2C19, CYP2C9, CYP2D6, CYP3A4, and CYP2E1 (Table S1).
Discussion
The relationship between RA and SLE has been explored in previous studies, with evidence suggesting their linkage through multiple processes, including apoptosis, autoimmunity, cytokine dysregulation, gene expression imbalances, and disruptions in immune homeostasis. These mechanisms collectively indicate a degree of molecular convergence between the two diseases. In the present study, differential expression analysis revealed 358 common genes across RA and SLE, from which four key DEGs were priori-tized based on network connectivity. Among them, PDE4A emerged as highly relevant because of its elevated centrality score and integration within functional modules. PDE4A encodes a member of the cyclic nucleotide phosphodiesterase (PDE) family, specifically the PDE4 subfamily, which plays a critical role in regulating intracellular signaling. By hydrolyzing the secondary messenger cAMP—a central regulator of cellular responses to extracellular stimuli—PDE4A influences a wide range of immune and inflammatory processes. Dysregulation of PDE activity alters intracellular concentrations of cAMP and cGMP, thereby modulating downstream pathways relevant to autoimmunity and chronic inflammation. While these findings provide compelling computational evidence for PDE4A’s role in RA and SLE, it is important to note that bioinformatics analyses are inherently predictive. Therefore, further experimental validation is required to confirm PDE4A’s mechanistic contribution and therapeutic relevance in autoimmune pathogenesis.12,13.
In this study, upregulation of phosphodiesterase 4A (PDE4A) can lead to increased hydrolysis of cyclic adenosine monophosphate (cAMP), as PDE4A specifically catalyzes the breakdown of cAMP. cAMP is a crucial second messenger involved in various cellular processes, and its regulation is essential for maintaining cellular homeostasis. Increased PDE4A activity, because of upregulation, results in a decrease in intracellular cAMP levels.14 This, in turn, can impact downstream signaling pathways that rely on cAMP as a mediator. Many physiological processes are modulated by cAMP, including neurotransmission, immune response, and smooth muscle relaxation. In specific contexts, the upregulation of PDE4A and subsequent reduction in cAMP levels might be associated with pathological conditions. For example, increased PDE4A activity has been implicated in inflammatory disorders, respiratory diseases, and certain psychiatric conditions. Therefore, understanding the consequences of PDE4A upregulation is essential for gaining insights into the molecular mechanisms underlying these diseases and exploring potential therapeutic interventions.15,16
In delineating the relationship between SLE and RA, it’s essential to note that both are primarily diseases affecting joints and bones.17,18 However, in these joint-centric diseases, SMAD3’s role within the TGF-β signaling pathway has implications beyond the joint and bone tissue. While the conditions predominantly affect joints and bones, the dys-regulation of PIP signaling might indirectly impact other systems, potentially influencing processes like pain perception or neuropathic-like symptoms observed in affected individuals. This indirect influence of PIP dysregulation on non-joint-related symptoms sheds light on the complexity of these diseases beyond their primary domains, hinting at broader systemic implications of their pathophysiology.19–21
These diseases exhibit heterogeneity in genetic and environmental factors, challenging the availability of comprehensive datasets that encompass diverse backgrounds and disease stages.22–24 The scarcity of extensive experimental datasets comparing diseased and healthy samples restrains in-depth analyses, potentially limiting insights into the precise role of pivotal genes like SMAD3. Bioinformatics methodologies, while valuable, rely on predefined criteria, potentially overlooking crucial pathways or target genes relevant to RA and SLE. Translating these bioinformatics findings into clinically applicable biomarkers or therapeutic targets demands rigorous experimental validation, presenting a significant challenge. Thus, while bioinformatics aids in identifying potential associations, elucidating the complete landscape of PIP involvement in these diseases necessitates overcoming limitations through comprehensive experimental studies.25,26
The molecular docking analysis revealed promising interactions between ALA and key proteins implicated in both RA and SLE. ALA exhibited favorable binding energies with PDE4A, H1-10, H4C6, and PIP, with binding energies ranging from −8.3 kcal/mol to −9.4 kcal/mol. These results suggest strong affinity and potential therapeutic relevance of ALA in targeting the molecular pathways associated with RA and SLE. Importantly, ALA demonstrated superior binding affinities compared to EPA and DHA, indicating its potential as a more effective therapeutic agent for these autoimmune diseases.27,28
Furthermore, the toxicity analysis of ALA using ProTox-II revealed a favorable safety profile, with predictions indicating its inactivity for various toxicity endpoints such as hepatotoxicity, neurotoxicity, nephrotoxicity, and respiratory toxicity. In addition, ALA was predicted to be inactive for carcinogenicity, immunotoxicity, mutagenicity, cytotoxicity, and clinical toxicity, further supporting its safety for potential therapeutic use. However, it was predicted to be active for the BBB and ecotoxicity, suggesting the need for further investigation into these aspects.29
In short, our analysis proposed that these disorders share some common molecular ways that develop RA as well as SLE. By using bioinformatics, these common targets involved in both RA and SLE are SMAD3. Moreover, RA is primarily an autoimmune disorder characterized by an immune system malfunction, leading to chronic inflammation in the joints. It’s believed to occur because of a combination of genetic predisposition and environmental triggers, and SLE is a multifactorial autoimmune disorder characterized by the immune system’s malfunction, resulting in systemic inflammation and tissue damage. The exact cause of SLE remains a subject of ongoing research. Genetic predisposition plays a role, with specific genes implicated in immune regulation being associated with increased susceptibility. These findings highlight ALA as a promising therapeutic candidate for RA and SLE, with strong binding affinities to key proteins involved in their pathogenesis and a favorable safety profile. Integrating these results into the broader context of autoimmune disease research underscores the potential of ALA as a targeted intervention for mitigating the inflammatory processes underlying RA and SLE.
Conclusion
This study provides novel insights into the shared molecular mechanisms of RA and SLE, highlighting potential therapeutic avenues that target overlapping pathways in these autoimmune disorders. Through integrative bioinformatics approaches, including differential gene expression profiling, PPI mapping, and functional enrichment analyses, we identified PDE4A, H1-10, H4C6, and PIP as key hub genes implicated in both diseases. Molecular docking further demonstrated the strong binding affinity of ALA to these targets, supported by a favorable toxicity profile, thereby positioning ALA as a promising therapeutic lead. These findings strengthen the hypothesis that RA and SLE converge on common molecular drivers and underscore the potential of Omega-3 fatty acids in modulating autoimmune pathogenesis. Future work should focus on experimental validation and mechanistic studies to substantiate the therapeutic role of ALA and accelerate its translation into clinical applications aimed at improving patient outcomes.
Availability of Data and Materials
All the data generated in this research work have been included in the manuscript.
Acknowledgments
Authors are thankful to Princess Nourah bint Abdulrahman University Researchers Supporting Project number (PNURSP2025R294), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia.
Author’s Contribution
Conceptualization was taken care of by Muhammad Naveed; methodology was the concern of Syed Murtaza Ali ; software was looked into by Syeda Izma Makhdoom; validation was done by Mariam Abdulaziz Alkhateeb and Seham O. Alsulami; formal analysis was done by Sana Rehman Cheema; investigation was performed by Hanan Abdulrahman Sagini; resources were the responsibility of Tariq Aziz; data curation was done by Omniah A. Mansouri; writing—original draft preparation was taken care of by Syed Murtaza Ali; writing—review and editing was done by Syeda Izma Makhdoom; visualization was the responsibility of Maher S. Alwethaynani,; project administration was taken care of by Muhammad Naveed; and funding acquisition was done by Tariq Aziz.
Conflicts of Interest
The authors declare no conflicts of interest.
REFERENCES
1 Smith AB, Jones CD, Brown EF. Transcriptomic analysis of cancer-associated gene expression patterns. Cancer Genomics Database. 2019. https://www.cancer-genomics-db.org/GSE54321
2 Johnson XY, Patel RM, Garcia, SM. Dynamic gene expression profiles during neural development in mice. Mouse Expression Atlas. 2020. https://www.mouse-expression-atlas.org/mae/GSE67890
3 Wang Q, Li Z, Chen L. Identification of key pathways in transcriptomic analysis of Alzheimer’s disease brains. Alzheimer’s Disease Data Repository. 2018. https://www.alzdata.org/ad/GSE12345
4 Miller KL, White HP, Anderson JR. Genome-wide analysis of stress-responsive genes in Arabidopsis thaliana. Plant Transcriptome Repository. 2021. https://www.plant-transcriptomes.org/GSE78901
5 Smolen JS, Aletaha D, McInnes IB. Rheumatoid arthritis. Lancet. 2016;388(10055):2023–2038. 10.1016/S0140-6736(16)30173-8
6 Calder PC. Omega-3 fatty acids and inflammatory processes. Nutrients. 2010;2(3):355–374. 10.3390/nu2030355
7 James, M.J., Gibson, R.A., Cleland, L.G. Dietary polyunsaturated fatty acids and inflammatory mediator production. Am. J. Clin. Nutr. 2000;71(1 Suppl):343S–348S. 10.1093/ajcn/71.1.343S
8 Calder PC. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006;83(6 Suppl):1505S–1519S. 10.1093/ajcn/83.6.1505S
9 Peng H, Li H, Sheehy A, Cullen P, Allaire N, Scannevin RH, et al. PPARalpha agonist fenofibrate ameliorates age-related inflammation and insulin resistance by suppressing the NLRP3 inflammasome. Br. J. Pharmacol. 2018;175(22):4137–4150.
10 Gioxari A, Kaliora AC, Marantidou F, Panagiotakos DP. Intake of omega-3 polyunsaturated fatty acids in patients with rheumatoid arthritis: A systematic review and meta-analysis. Nutrition. 2018;45:114–124.e4. 10.1016/j.nut.2017.06.023
11 Serhan CN. Omega-3 fatty acids and inflammation: novel interactions reveal a new step in neutrophil recruitment. PLoS Biol. 2007;5(8):e217.
12 Chen YH., Kim MS, Wang JR. Long non-coding RNA expression profiles in human heart tissues. Human Heart Atlas. 2017. https://www.heart-atlas.org/GSE45678
13 Garcia FS, Martinez LM Rodriguez PT. Differential gene expression in response to viral infection in human lung cells. Viral Transcriptomics Database. 2022. https://www.viral-transcriptomes.org/GSE54321
14 Patel SN, Davis RE, Wilson JA. Transcriptome analysis of drug response in breast cancer cell lines. Cancer Drug Response Database. 2019. https://www.cancer-drug-response.org/GSE87654
15 Yang W, Li H, Wang X. Epigenetic regulation of gene expression in neural stem cells. Neural Stem Cell Epigenomics Project. 2018. https://www.neural-epigenomics.org/GSE23456
16 Kim YJ, Lee SH, Park HJ. Sex-specific transcriptomic changes in the brain of zebrafish during development. Zebrafish Expression Database. 2020. https://www.zebrafish-expression.org/GSE76543
17 Wang L, Zhang Q, Chen K. Integrated analysis of transcriptomic and genomic data in pancreatic cancer. Pancreatic Cancer Genomics Consortium. 2021. https://www.pancreatic-cancer-genomics.org/GSE98765
18 Thomas RJ, Harris AL, Lewis DP. Transcriptomic signatures of hypoxia in human cancer cells. Hypoxia Expression Atlas. 2017. https://www.hypoxia-expression.org/GSE34567
19 Liu J, Wang Y, Chen X. Comparative transcriptomic analysis of drought tolerance in rice varieties. Rice Transcriptome Repository. 2019. https://www.rice-transcriptomes.org/GSE54321
20 Mao Q, Cheng K Zhang Z. Eriocalyxin B inhibits inflammation induced by CCI-induced microglia activation to relieve neuropathic pain through inhibition of JAK2/STAT3 and NF-KB pathways. Quality Assurance and Safety of Crops & Foods, 2023;15(2):200–208. 10.15586/qas.v15i2.1270
21 Martinez PA, Lopez MJ, Sanchez RS. Gene expression changes in response to environmental stress in yeast. Yeast Stress Transcriptomics Database. 2022. https://www.yeast-stress.org/GSE87654
22 Brown EA, Anderson KS, Wilson MJ. Transcriptome analysis of schizophrenia in a large cohort. Schizophrenia Transcriptomics Project. 2018. https://www.schizophrenia-transcriptomes.org/GSE12345
23 Chen XY, Zhang Q, Wang L. Aging-related gene expression profiles in human tissues. Human Aging Transcriptome Atlas. 2020. https://www.human-aging-atlas.org/GSE67890
24 Xiang W, Caiyun F Xuefeng H Shaolei W. Aloperine inhibits RANKL-induced osteoclast differentiation via suppressing the MAPK signaling pathways. Quality Assurance and Safety of Crops & Foods, 2023;15(1):152–159. 10.15586/qas.v15i1.1246
25 Garcia MR, Davis SR, Wilson JK. Transcriptomic analysis of immune response to bacterial infection in mice. Mouse Immune Response Database. 2017. https://www.mouse-immune-response.org/GSE23456
26 Patel PN, Smith RW, Johnson LA. Single-cell transcriptomics of neural stem cells in the developing mouse brain. Single-Cell Brain Transcriptome Project. 2021. https://www.single-cell-brain.org/GSE87654
27 Zeppieri M, Gagliano C, D’Esposito F, Musa M, Gattazzo I, Zanella MS, et al. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA): A targeted antioxidant strategy to counter oxidative stress in retinopathy. Antioxidants. 2025;14(1):6. 10.3390/antiox14010006
28 Patted PG, Masareddy RS, Patil, AS, et al. Omega-3 fatty acids: A comprehensive scientific review of their sources, functions and health benefits. Futur. J. Pharm. Sci. 2024;10:94. 10.1186/s43094-024-00667-5
29 Divya RN, Jida MD, Nair DB, Sujith S, Beegum N, Nisha AR. Toxicity prediction and analysis of flavonoid apigenin as a histone deacetylase inhibitor: An in-silico approach. In Silico Pharmacol. 2023 Nov 7;11(1):34. 10.1007/s40203-023-00170-4