
Download
REVIEW ARTICLE
Chronic HBV infection, stemness pathways, and compromised immune surveillance in HCC development
Negin Razaghia, Ashraf Kariminikb*, Mehdi Ranjbarc, Morteza Bahaaldin-beygib
aDepartment of Pharmacology and Toxicology, School of Pharmacy, Kerman University of Medical Sciences, Kerman, Iran
bDepartment of Microbiology, Ke.C., Islamic Azad University, Kerman, Iran
cPharmaceutics Research Center, Institute of Neuropharmacology, Kerman University of Medical Sciences, Kerman, Iran
Abstract
Chronic hepatitis B virus (HBV) infection is a leading cause of hepatocellular carcinoma (HCC). This review elucidates the molecular pathways by which HBV promotes the expression of the stemness transcription factors OCT4, Sox2, and NANOG. Through a comprehensive literature review, we found that HBV enhances their expression via multiple mechanisms. These include HBxAg-induced chromatin remodeling, activation of histone demethylase KDM5B, and integration of the truncated HBx-ΔC protein into the host genome. Furthermore, chronic inflammation driven by persistent HBV infection acts as a key driver for their overexpression. This dysregulation is strongly associated with increased tumor proliferation, metastasis, and poor prognosis in HBV-infected HCC patients. Consequently, OCT4, Sox2, and NANOG emerge as promising biomarkers for early detection and prognosis, as well as potential therapeutic targets. Modulating their activity could offer novel strategies for targeted treatment. In conclusion, HBV-induced alterations in these transcription factors represent critical oncogenic mechanisms in HCC. Further research is essential to develop novel therapeutic approaches that regulate their activity, ultimately improving clinical outcomes for patients.
Key words: OCT4, NANOG, Sox2, Hepatocellular Carcinoma, Hepatitis B Virus
*Corresponding author: Ashraf Kariminik, Department of Microbiology, Ke.C., Islamic Azad University, Kerman, Iran. Email address: [email protected]
Received 8 May 2025; Accepted 22 October 2025; Available online 1 May 2026
Copyright: Razaghi N, et al.
This open access article is licensed under Creative Commons Attribution 4.0 International (CC BY 4.0). http://creativecommons.org/licenses/by/4.0/
Introduction
Hepatocellular carcinoma (HCC) is one of the most prevalent and lethal malignancies worldwide, ranking among the leading causes of cancer-related mortality.1 The incidence of HCC is particularly high in regions where chronic hepatitis B virus (HBV) infection is endemic, underscoring the strong etiological link between HBV and liver cancer development.2 Despite advancements in diagnostic and therapeutic strategies, the prognosis for HCC remains poor, largely due to its aggressive nature, late-stage diagnosis, and limited treatment options.3 Chronic HBV infection is a primary risk factor for HCC. It contributes to oncogenesis through a complex interplay of genetic, epigenetic, and immunological mechanisms. Key events include the integration of the HBV genome into host DNA, which cause genomic instability, activate oncogenes (e.g., TERT), and disrupt tumor suppressor genes (e.g., p53).5 Persistent HBV infection triggers chronic liver inflammation. This inflammatory state induces oxidative stress, continuous hepatocyte damage, and the creation of a protumorigenic microenvironment, all of which accelerate disease progression.6 Recent research has highlighted the crucial role of cancer stem cells (CSCs) in HBV-associated hepatocarcinogenesis. CSCs are a subpopulation of tumor cells with self-renewal capacity, high proliferative potential, and resistance to conventional therapies, making them central to tumor initiation, maintenance, and recurrence.7 Among the key regulators of CSC properties are the transcription factors OCT4, Sox2, and NANOG.8 While essential for maintaining embryonic stem cell pluripotency, their aberrant expression in cancers, including HBV-related HCC, is linked to enhanced tumorigenicity, tumor heterogeneity, and therapy resistance.9,10 This review provides a comprehensive analysis of the involvement of OCT4, Sox2, and NANOG in HBV-related HCC. We focus on their molecular mechanisms of regulation, the signaling pathways they engage with (e.g., Wnt/β-catenin, Notch),11 and their potential as therapeutic targets. Our goal is to shed light on CSC regulation in HBV-driven liver cancer and discuss strategies for targeted clinical interventions.
Direct Oncogenic Mechanisms of Hepatitis B Virus
Hepatitis B virus exerts direct oncogenic effects through the actions of its viral genome and encoded proteins, which subvert fundamental cellular processes to drive malignant transformation.12 A primary mechanism is the integration of HBV-DNA into the host hepatocyte genome. Although dispensable for viral replication, this integration occurs with high frequency in chronic infection and serves as a potent driver of genomic instability.13 Integrated viral sequences can disrupt tumor suppressor loci such as TP53 and RB1 leading to loss of cell cycle control and unchecked proliferation.14 Conversely, integration events frequently activate proto-oncogenes; for instance, insertional mutagenesis near the TERT promoter is a recurrent event in HBV-associated HCC, resulting in telomerase reactivation and cellular immortalization. Furthermore, HBV integration induces large-scale chromosomal rearrangements, deletions, and translocations, collectively fostering a mutagenic landscape conducive to carcinogenesis.15 The HBx protein, a multifunctional regulatory protein, is a central oncogenic effector. HBx lacks intrinsic DNA-binding capacity but functions as a transcriptional coactivator by interacting with key cellular factors, including p53, NF-κB, AP-1, and SP1. Through these interactions, HBx reprograms host gene expression networks to favor proliferation and survival. It directly inhibits p53-mediated apoptosis, enabling the persistence of genetically damaged cells.16 At the epigenetic level, HBx recruits chromatin-modifying complexes to specific genomic loci, altering histone posttranslational modifications and DNA methylation patterns to silence tumor suppressors or activate oncogenes. Critically, HBx activates the canonical Wnt/β-catenin pathway by stabilizing β-catenin, thereby promoting hepatocyte dedifferentiation, acquisition of stem-like properties, and tumor initiation. Additionally, HBx compromises genomic integrity by suppressing nucleotide excision repair (NER) and base excision repair (BER) pathways, leading to the accumulation of unrepaired DNA damage.17 The HBV envelope proteins, particularly the large surface protein (LHBs) containing the PreS1/PreS2 domains, and also contribute directly to oncogenesis. When overexpressed as commonly occurs in chronic infection, these proteins accumulate in the endoplasmic reticulum (ER), inducing severe ER stress and activating the unfolded protein response (UPR). Sustained UPR signaling shifts the cellular balance from proapoptotic to prosurvival outcomes, conferring resistance to cell death. ER stress further generates reactive oxygen species (ROS), causing oxidative DNA damage and genomic instability.18 Concurrently, PreS/S proteins activate mitogenic signaling cascades, including the MAPK/ERK and PI3K/AKT pathways, which synergistically promote hepatocyte proliferation, survival, and malignant transformation.19
Indirect Oncogenic Mechanisms of Hepatitis B Virus
Beyond direct viral interference, HBV promotes hepatocarcinogenesis through chronic immune-mediated inflammation and microenvironmental remodeling.20 Persistent viral infection triggers a sustained, yet ineffective, host immune response characterized by the continuous recruitment and activation of immune cells within the liver parenchyma. This state of chronic inflammation is marked by elevated levels of proinflammatory cytokines, including tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), and interferon-gamma (IFN-γ). These cytokines induce oxidative stress via the generation of reactive oxygen and nitrogen species (ROS/RNS), which directly damage DNA, proteins, and lipids, accelerating the acquisition of oncogenic mutations.21 A critical consequence of persistent antigen exposure is immune exhaustion. Cytotoxic T lymphocytes (CTLs) and natural killer (NK) cells, essential for tumor immune surveillance, become functionally impaired or depleted.21 This collapse in adaptive and innate immunity permits the survival and clonal expansion of premalignant hepatocytes harboring oncogenic alterations. Concurrently, chronic inflammation drives progressive hepatic fibrosis, culminating in cirrhosis, a well-established premalignant condition. The fibrotic microenvironment, rich in activated hepatic stellate cells and remodeled extracellular matrix, provides a protumorigenic niche that supports angiogenesis, cell survival, and invasion. HBV also indirectly fuels carcinogenesis by dysregulating the hepatic progenitor cell (HPC) compartment.22 Chronic injury and inflammation stimulate HPC proliferation as a compensatory regenerative mechanism. However, HBV subverts this process by modulating key stemness-associated signaling pathways, notably Wnt/β-catenin and Notch, which are crucial for HPC self-renewal and fate determination. This dysregulation, often mediated by inflammatory cytokines like IL-6, leads to the pathological overexpression of pluripotency transcription factors such as OCT4, SOX2, and NANOG.23 Consequently, HPCs or transdifferentiated hepatocytes acquire cancer stem cell (CSC)-like properties, including enhanced self-renewal, therapy resistance, and tumor-initiating capacity. Epigenetic modifications, including aberrant DNA methylation and histone modifications induced by the inflammatory milieu, further lock these cells into a malignant, undifferentiated state, thereby bridging chronic inflammation to the emergence of HCC.24
The Biology of OCT4, Sox2, and NANOG
OCT4, Sox2, and NANOG are master transcription factors essential for maintaining the self-renewal and pluripotency of embryonic stem cells. They function by binding to specific promoter and enhancer regions (e.g., the octamer motif ATGCAAAT for OCT4) to regulate the expression of downstream target.25 These factors are encoded by the Pou5f1 (OCT4), Sox2, and NANOG genes, located on human chromosomes 17, 3, and 12, respectively. Their expression is tightly controlled through a combination of transcriptional, posttranslational (e.g., phosphorylation, ubiquitination), and epigenetic mechanisms.26,27 The transcriptional network governed by OCT4, SOX2, and NANOG is central to the maintenance of pluripotency in embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). Beyond their canonical role in self-renewal and lineage restriction, these core pluripotency factors are critically implicated in the acquisition and stabilization of cancer stem cell (CSC) phenotypes in hepatocellular carcinoma (HCC).28 Their aberrant reexpression in somatic tissues, particularly in the context of chronic liver injury or viral hepatitis, facilitates cellular reprograming toward an undifferentiated, tumor-initiating state. A defining feature of this regulatory axis is its exquisite dosage sensitivity. Precise stoichiometric levels of OCT4, SOX2, and NANOG are essential for preserving pluripotency; even minor deviations can trigger lineage commitment or differentiation.29 For instance, elevated OCT4 expression promotes self-renewal, whereas its downregulation is necessary for endodermal or mesodermal specification.26 This dose-dependent functionality is mechanistically underpinned by the formation of context-specific transcriptional complexes: varying concentrations of OCT4, for example, determine its binding partners (e.g., SOX2, KLF4, or lineage-specific factors), thereby modulating enhancer occupancy and target gene expression profiles. Post-translational modifications (PTMs) serve as critical rheostats for fine-tuning the activity, stability, and subcellular localization of these factors. Phosphorylation events, often mediated by kinases such as AKT or ERK, enhance transcriptional activity and protein stability, whereas ubiquitination catalyzed by E3 ligases such as WWP2 or FBXW8 targets these proteins for proteasomal degradation, thereby attenuating their function.26 This dynamic regulation allows rapid adaptation to extracellular cues and intracellular signaling states.
Role of OCT4, SOX2, and NANOG in Cancer and Liver Disease
The functional output of OCT4, SOX2, and NANOG is profoundly influenced by crosstalk with key developmental and oncogenic signaling pathways. Notably, transforming growth factor-beta (TGF-β) signaling can either reinforce or antagonize pluripotency depending on cellular context and duration of activation. In early hepatocarcinogenesis, TGF-β may promote epithelial-mesenchymal transition (EMT) and CSC expansion via synergistic upregulation of NANOG and SOX2, thereby linking microenvironmental stress to stemness reprograming.30 Importantly, the pathogenic relevance of these factors extends beyond HCC. In chronic liver diseases such as cirrhosis, OCT4 and NANOG are reactivated in hepatic progenitor cells, contributing to ductular reactions and fibrogenic responses. This reprograming is markedly amplified in the setting of viral coinfection; for example, concurrent hepatitis C virus (HCV) infection synergizes with HBV to potentiate TGF-β1–driven OCT4/NANOG expression, accelerating fibrosis and increasing malignant transformation risk.31 Thus, OCT4, SOX2, and NANOG function not merely as static markers but as dynamic integrators of oncogenic, inflammatory, and regenerative signals in the liver.
Molecular Drivers of OCT4, SOX2, and NANOG Dysregulation in HBV-Associated HCC
A robust body of evidence positions the pathological overexpression of the core pluripotency transcription factors OCT4, SOX2, and NANOG as a molecular hallmark of HBV-driven HCC. This dysregulation is not merely an epiphenomenon but is mechanistically driven by specific viral components and host–virus interactions, directly linking it to oncogenesis and adverse clinical outcomes. Clinically, studies consistently report significant upregulation of these factors in HBV-HCC tumor tissues compared to adjacent nontumorous parenchyma. For instance, Ye et al. demonstrated that elevated OCT4 expression in HBV-derived tumors strongly correlates with the transcriptional dysregulation of downstream oncogenic targets, including thymidine kinase 1 (TK1) and cortactin (CTTN), and serves as an independent predictor of reduced overall survival.9 This overexpression is actively orchestrated by HBV oncoproteins. The PreS1 envelope protein, for example, has been shown by Liu et al. to induce the expression of canonical cancer stem cell (CSC) markers (CD133, CD90) while simultaneously upregulating OCT4 and NANOG, thereby conferring enhanced tumorigenic potential upon both normal hepatocytes and hepatoma cell lines in murine xenograft models.32 Similarly, the multifunctional HBx protein acts as a central node in this regulatory network.33 Arzumanyan et al. and Wang et al. established that HBx drives hepatic carcinogenesis partly through the activation of the histone demethylase KDM5B, which epigenetically reprograms chromatin to facilitate the transcriptional upregulation of OCT4 and NANOG, thereby inducing stem cell-like properties in infected hepatocytes.34,35 This oncogenic potential is further amplified by the integration of C-terminally truncated HBx (HBx-ΔC) into the host genome. As demonstrated by Ching et al., cells harboring HBx-ΔC exhibit significantly enhanced tumorigenicity, migratory capacity, and expression of CSC markers and pluripotency factors compared to those expressing full-length HBx or vector controls, a finding corroborated in clinical HCC tissue samples.31,36 Beyond direct viral induction, cellular reprograming events also contribute. Epithelial cell adhesion molecule (EpCAM), normally silenced in mature hepatocytes by Polycomb Repressive Complex 2 (PRC2), is frequently re-expressed in HBV-HCC due to PRC2 dysfunction. Mani et al. provided compelling evidence that EpCAM re-expression is not a passive consequence but an active driver, directly upregulating OCT4, SOX2, and NANOG in both in vitro and in vivo settings.37 Conversely, the tumor-suppressive factor ATOH8, which is downregulated in nearly half of all HBV-related HCC cases, acts as a critical negative regulator. Song et al. reported a strong inverse correlation between ATOH8 levels and those of OCT4/SOX2/NANOG, and crucially, demonstrated that restoring ATOH8 expression suppresses these pluripotency factors and inhibits tumor progression.38
The Inflammatory Microenvironment as a Potent Amplifier of Stemness
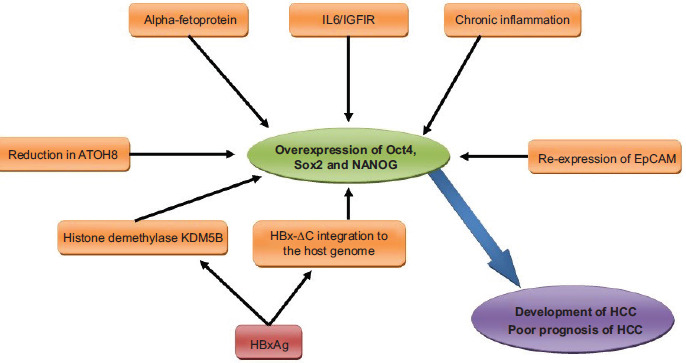
Chronic inflammation, a defining feature of persistent HBV infection, serves as a powerful environmental amplifier of the OCT4/SOX2/NANOG axis, directly linking immune dysregulation to poor prognosis. Chang et al. elucidated a key mechanistic pathway wherein activation of the interleukin-6 (IL-6)/insulin-like growth factor-I receptor (IGF-1R) signaling cascade potently induces the expression of OCT4 and NANOG, thereby establishing a direct molecular conduit from the inflammatory tumor microenvironment to the acquisition of stemness and aggressive tumor behavior.39 This is consistent with findings that chronic inflammation itself is a primary driver of HCC initiation in HBV-infected individuals, with multiple studies confirming its role in elevating OCT4 and NANOG expression within the infected liver tissue.40–42 The functional significance of this overexpression is underscored by rigorous experimental validation. Xu et al. conducted a seminal study comparing two clonally related cell lines, Hep-11 (derived from a primary HCC) and Hep-12 (derived from a recurrent tumor in the same patient), which shared identical HBV integration sites. Despite their genetic kinship, the Hep-12 line exhibited markedly higher expression of liver CSC markers and the transcription factors OCT4, SOX2, and NANOG. Strikingly, Hep-12 cells demonstrated a tenfold greater tumorigenic potential in immunodeficient mice compared to Hep-11 cells, providing direct in vivo evidence that elevated expression of these pluripotency factors is a primary determinant of enhanced tumor-initiating capacity and disease recurrence.43 This observation is further supported by clinical data indicating that sustained overexpression of OCT4, SOX2, and NANOG in HCC tissue is consistently associated with a poor prognosis in HBV-related cases.44 Collectively, these findings underscore that the functional duality of OCT4, SOX2, and NANOG in the liver, spanning roles in regeneration, antiviral defense, and oncogenesis, is critically governed by their expression levels. In the context of chronic hepatitis B infection, HBV orchestrates their pathological overexpression through a constellation of molecular mechanisms, comprehensively summarized in Figure 1.
Figure 1 Molecular mechanisms driving the overexpression of OCT4, SOX2, and NANOG in HBV-infected hepatocytes and hepatic progenitor cells. Chronic hepatitis B virus (HBV) infection induces the pathological upregulation of the pluripotency transcription factors OCT4, SOX2, and NANOG through a network of interconnected pathways. Key inducers include viral oncoproteins (notably HBx and its truncated form HBx-ΔC, which activates the histone demethylase KDM5B), the HBV envelope protein PreS1, the oncofetal protein alpha-fetoprotein (AFP), chronic inflammation and its associated IL-6/IGF-1R signaling, downregulation of the tumor suppressor ATOH8, and re-expression of Epithelial Cell Adhesion Molecule (EpCAM) following HBV-mediated loss of Polycomb Repressive Complex 2 (PRC2) function. The concerted action of these factors leads to sustained overexpression of OCT4, SOX2, and NANOG, which in turn drives the acquisition of cancer stem cell (CSC) properties, hepatocellular carcinoma (HCC) development, and is strongly associated with poor clinical prognosis.
Clinical Implications, Prognostic Value, and Context-dependent Biology
The dysregulation of OCT4, SOX2, and NANOG carries significant clinical implications, positioning them as valuable prognostic biomarkers and potential therapeutic targets. A meta-analysis by Liang et al. confirmed that high OCT4 expression is significantly associated with larger tumor size, increased tumor number, and advanced differentiation stages in HCC, solidifying its utility as a prognostic indicator.29 Moreover, the coexpression of these transcription factors predicts an even worse clinical outcome for patients with HBV-related HCC, suggesting synergistic oncogenic effects.45 However, their role is multifaceted and context-dependent, necessitating a nuanced understanding. Host genetic and epigenetic factors significantly modulate their function. For instance, Fan et al. revealed that DNA methylation at specific target sites can enhance the oncogenic activity of these factors, effectively directing hepatic progenitor cells toward a malignant CSC fate.46 Furthermore, polymorphisms within the OCT4 gene have been linked to an increased susceptibility to chronic hepatitis B and subsequent HCC development, highlighting the interplay between host genetics and viral pathogenesis.47 The regulatory landscape is further complicated by noncoding RNAs; emerging evidence indicates that specific microRNAs play crucial roles in the pathogenesis of HBV-related HCC, though their precise interactions with the OCT4/SOX2/NANOG axis remain an area ripe for future investigation.48 Critically, the temporal dynamics of infection must be considered. Bautista et al. demonstrated that short-term HBV infection of hepatic stem or progenitor cells in vitro does not alter OCT4 or NANOG expression, suggesting that prolonged infection and the resultant chronic inflammation are prerequisites for their pathological overexpression.49 Finally, it is imperative to acknowledge the dual nature of these factors. While this review focuses on their tumorigenic roles in the context of chronic HBV, evidence indicates they also possess protective functions. For example, Yang et al. showed that SOX2 can directly bind to the HBV enhancer II/core promoter region and repress viral replication, suggesting an intrinsic antiviral role.50 Therefore, the ultimate biological outcome, whether regenerative, antiviral, or oncogenic, is determined by the duration of infection, the specific viral and host factors involved, and the broader cellular and inflammatory context. This duality presents both a challenge and an opportunity for developing targeted therapies that selectively inhibit their oncogenic functions without compromising their physiological roles in liver homeostasis and repair.
Conclusions
This review establishes that HBV-driven hepatocellular carcinoma is critically dependent on the pathological reactivation of the pluripotency factors OCT4, SOX2, and NANOG. Through viral proteins (HBx, PreS1, HBx-ΔC), epigenetic modifiers (KDM5B, PRC2 loss), and chronic inflammation (IL-6/IGF-1R, Wnt/β-catenin), HBV hijacks stemness pathways to generate and sustain cancer stem cells, driving tumor initiation, metastasis, and therapy resistance. While these factors represent promising prognostic biomarkers and therapeutic targets, their dual roles in liver regeneration and intrinsic antiviral defense exemplified by SOX2’s ability to suppress HBV replication, demand highly selective targeting strategies to avoid compromising tissue homeostasis. Critical knowledge gaps remain: the regulatory networks involving microRNAs and chromatin modifiers, the impact of host genetic variants (e.g., OCT4 polymorphisms), and the precise temporal dynamics of their dysregulation during chronic infection. Addressing these through integrated molecular and clinical studies will be essential for developing context-specific interventions that improve outcomes in HBV-HCC.
Mandatory Disclosure on Use of Artificial Intelligence
The authors declare that no AI-assisted tools were used in the preparation of this manuscript. All references have been manually verified for accuracy and relevance.
Authors Contributions
All authors contributed equally to the manuscript preparation.
Conflict of Interest
The authors declare no conflict of interest.
Funding
None.
REFERENCES
1 Naghib M, Kariminik A, Kazemi Arababadi M. TLR2, as a pathogen recognition receptor, plays critical roles in hepatitis B outcome. Viral Immunol. 2022;35(1):15–23. 10.1089/vim.2021.0141
2 Nahavandi-Parizi P, Kariminik A, Montazeri M. Retinoic acid-inducible gene 1 (RIG-1) and IFN-β promoter stimulator-1 (IPS-1) significantly down-regulated in the severe coronavirus disease 2019 (COVID-19). Mol Biol Rep. 2023;50(1):907–11. 10.1007/s11033-022-07981-2
3 Brown ZJ, Tsilimigras DI, Ruff SM, Mohseni A, Kamel IR, Cloyd JM, et al. Management of hepatocellular carcinoma: A review. JAMA Surg. 2023;158(4):410–20. 10.1001/jamasurg.2022.7989
4 Capasso M, Cossiga V, Guarino M, Ranieri L, Morisco F. The role of hepatitis viruses as drivers of hepatocancerogenesis. Cancers. 2024;16(8):1505. 10.3390/cancers16081505
5 Bahaaldin-Beygi M, Kariminik A, Arababadi MK. Royal jelly significantly alters inflammasome pathways in patients with chronic hepatitis B. IJEB. 2022;60(11):875–9. 10.56042/ijeb.v60i11.60264
6 Zhang M, Chen H, Liu H, Tang H. The impact of integrated hepatitis B virus DNA on oncogenesis and antiviral therapy. Biomark Res. 2024;12(1):84. 10.1186/s40364-024-00611-y
7 Zhou L, He L, Liu CH, Qiu H, Zheng L, Sample KM, et al. Liver cancer stem cell dissemination and metastasis: Uncovering the role of NRCAM in hepatocellular carcinoma. J Exp Clin Cancer Res. 2023;42(1):311. 10.1186/s13046-023-02893-w
8 Roy A, Mishra J, Chakraborty S, Singh SP, Patra SK. Epigenetic regulation of pluripotency inducer genes NANOG and SOX2 in human prostate cancer. Progress Mol Biol Transl Sci. 2023; 197:241–60. 10.1016/bs.pmbts.2023.01.010
9 Ye C, Zhang X, Chen X, Cao Q, Zhang X, Zhou Y, et al. Multiple novel hepatocellular carcinoma signature genes are commonly controlled by the master pluripotency factor OCT4. Cell Oncol. 2020; 43:279–95. 10.1007/s13402-019-00487-3
10 Ching RH, Sze KM, Lau EY, Chiu YT, Lee JM, Ng IO, et al. C-terminal truncated hepatitis B virus X protein regulates tumorigenicity, self-renewal and drug resistance via STAT3/Nanog signaling pathway. Oncotarget. 2017;8(14):23507. 10.18632/oncotarget.15183
11 Martins-Neves SR, Sampaio-Ribeiro G, Gomes CM. Self-renewal and pluripotency in osteosarcoma stem cells’ chemoresistance: Notch, hedgehog, and wnt/β-catenin interplay with embryonic markers. Int J Mol Sci. 2023;24(9): 8401. 10.3390/ijms24098401
12 Borgia M, Dal Bo M, Toffoli G. Role of virus-related chronic inflammation and mechanisms of cancer immune-suppression in pathogenesis and progression of hepatocellular carcinoma. Cancers. 2021;13(17):4387. 10.3390/cancers13174387
13 Tu T, Budzinska MA, Shackel NA, Urban S. HBV DNA integration: Molecular mechanisms and clinical implications. Viruses. 2017;9(4):75. 10.3390/v9040075
14 Li W, Wang S, Jin Y, Mu X, Guo Z, Qiao S, et al. The role of the hepatitis B virus genome and its integration in the hepatocellular carcinoma. Front Microbiol. 2024:15:1469016. 10.3389/fmicb.2024.1469016
15 Sze KM, Ho DW, Chiu YT, Tsui YM, Chan LK, Lee JM, et al. Hepatitis B virus–telomerase reverse transcriptase promoter integration harnesses host ELF4, resulting in telomerase reverse transcriptase gene transcription in hepatocellular carcinoma. Hepatology. 2021;73(1):23–40. 10.1002/hep.31231
16 Tarocchi M, Polvani S, Marroncini G, Galli A. Molecular mechanism of hepatitis B virus-induced hepatocarcinogenesis. World J Gastroenterol. 2014;20(33): 11630–40. 10.3748/wjg.v20.i33.11630
17 Gómez-Moreno A, Garaigorta U. Hepatitis B virus and DNA damage response: Interactions and consequences for the infection. Viruses. 2017;9(10):304. 10.3390/v9100304
18 Hu T, Wang J, Li W, Liu M, Han N, Yuan M, et al. Endoplasmic reticulum stress in hepatitis B virus and hepatitis C virus infection. Viruses. 2022;14(12):2630. 10.3390/v14122630
19 Shoraka S, Hosseinian SM, Hasibi A, Ghaemi A, Mohebbi SR. The role of hepatitis B virus genome variations in HBV-related HCC: Effects on host signaling pathways. Front Microbiol. 2023;14:1213145. 10.3389/fmicb.2023.1213145
20 D’souza S, Lau KC, Coffin CS, Patel TR. Molecular mechanisms of viral hepatitis induced hepatocellular carcinoma. World J Gastroenterol. 2020;26(38):5759–83. 10.3748/wjg.v26.i38.5759
21 Dimitriadis K, Katelani S, Pappa M, Fragkoulis GE, Androutsakos T. The role of interleukins in HBV infection: A narrative review. J Pers Med. 2023;13(12):1675. 10.3390/jpm13121675
22 Hou XJ, Ye F, Li XY, Liu WT, Jing YY, Han ZP, et al. Immune response involved in liver damage and the activation of hepatic progenitor cells during liver tumorigenesis.Cell Immunol. 2018;326:52–9. 10.1016/j.cellimm.2017.08.004
23 Daud M, Rana MA, Husnain T, Ijaz B. Modulation of Wnt signaling pathway by hepatitis B virus. Arch Virol. 2017;162(10):2937–47. 10.1007/s00705-017-3462-6
24 Wang R, Sun Q, Wang P, Liu M, Xiong S, Luo J, et al. Notch and Wnt/β-catenin signaling pathway play important roles in activating liver cancer stem cells. Oncotarget. 2016;7(5):5754–68. 10.18632/oncotarget.6805
25 Vasefifar P, Motafakkerazad R, Maleki LA, Najafi S, Ghrobaninezhad F, Najafzadeh B, et al. Nanog, as a key cancer stem cell marker in tumor progression. Gene. 2022;827:146448. 10.1016/j.gene.2022.146448
26 Sohn EJ, Moon HJ, Lim JK, Kim DS, Kim JH. Regulation of the protein stability and transcriptional activity of OCT4 in stem cells. Adv Biol Regul. 2021;79:100777. 10.1016/j.jbior.2020.100777
27 Rizzino A. Sox2 and Oct-3/4: A versatile pair of master regulators that orchestrate the self-renewal and pluripotency of embryonic stem cells. Wiley Interdiscip Rev Syst Biol Med. 2009;1(2):228–36. 10.1002/wsbm.12
28 Zheng YW, Nie YZ, Taniguchi H. Cellular reprogramming and hepatocellular carcinoma development. World J Gastroenterol. 2013;19(47):8850–60. 10.3748/wjg.v19.i47.8850
29 Liang C, Xu Y, Ge H, Li G, Wu J. Clinicopathological and prognostic significance of OCT4 in patients with hepatocellular carcinoma: A meta-analysis. Onco Targets Ther. 2018;11:47–57. 10.2147/OTT.S151390
30 Gordeeva O. TGFβ Family signaling pathways in pluripotent and teratocarcinoma stem cells’ fate decisions: Balancing between self-renewal, differentiation, and cancer. Cells. 2019;8(12). 10.3390/cells8121500
31 Li W, Duan X, Zhu C, Liu X, Jeyarajan AJ, Xu M, et al. Hepatitis B and hepatitis C virus infection promote liver fibrogenesis through a TGF-β1-Induced OCT4/Nanog pathway. J Immunol. 2022;208(3):672–84. 10.4049/jimmunol.2001453
32 Liu Z, Dai X, Wang T, Zhang C, Zhang W, Zhang W, et al. Hepatitis B virus PreS1 facilitates hepatocellular carcinoma development by promoting appearance and self-renewal of liver cancer stem cells. Cancer Lett. 2017;400:149–60. 10.1016/j.canlet.2017.04.017
33 Zhu M, Li W, Lu Y, Dong X, Lin B, Chen Y, et al. HBx drives alpha fetoprotein expression to promote initiation of liver cancer stem cells through activating PI3K/AKT signal pathway. Int J Cancer. 2017;140(6):1346–55. 10.1002/ijc.30553
34 Arzumanyan A, Friedman T, Ng IO, Clayton MM, Lian Z, Feitelson MA. Does the hepatitis B antigen HBx promote the appearance of liver cancer stem cells? Cancer Res. 2011;71(10):3701–8. 10.1158/0008-5472.CAN-10-3951
35 Wang X, Oishi N, Shimakami T, Yamashita T, Honda M, Murakami S, et al. Hepatitis B virus X protein induces hepatic stem cell-like features in hepatocellular carcinoma by activating KDM5B. World J Gastroenterol. 2017;23(18):3252–61. 10.3748/wjg.v23.i18.3252
36 Ching RH, Sze KM, Lau EY, Chiu YT, Lee JM, Ng IO, et al. C-terminal truncated hepatitis B virus X protein regulates tumorigenicity, self-renewal and drug resistance via STAT3/Nanog signaling pathway. Oncotarget. 2017;8(14):23507–16. 10.18632/oncotarget.15183
37 Mani SK, Zhang H, Diab A, Pascuzzi PE, Lefrançois L, Fares N, et al. EpCAM-regulated intramembrane proteolysis induces a cancer stem cell-like gene signature in hepatitis B virus-infected hepatocytes. J Hepatol. 2016;65(5):888–98. 10.1016/j.jhep.2016.05.022
38 Song Y, Pan G, Chen L, Ma S, Zeng T, Chan TH, et al. Loss of ATOH8 increases stem cell features of hepatocellular carcinoma cells. Gastroenterology. 2015;149(4):1068–81 e5. 10.1053/j.gastro.2015.06.010
39 Chang TS, Wu YC, Chi CC, Su WC, Chang PJ, Lee KF, et al. Activation of IL6/IGFIR confers poor prognosis of HBV-related hepatocellular carcinoma through induction of OCT4/NANOG expression. Clin Cancer Res. 2015;21(1):201–10. 10.1158/1078-0432.CCR-13-3274
40 Gufler S, Seeboeck R, Schatz C, Haybaeck J. The translational bridge between inflammation and hepatocarcinogenesis. Cells. 2022;11(3):533. 10.3390/cells11030533
41 Chang TS, Chen CL, Wu YC, Liu JJ, Kuo YC, Lee KF, et al. Inflammation promotes expression of stemness-related properties in HBV-related hepatocellular carcinoma. PLoS One. 2016;11(2):e0149897. 10.1371/journal.pone.0149897
42 Tsukamoto H, Mishra L, Machida K. Alcohol, TLR4-TGF-β antagonism, and liver cancer. Hepatol Int. 2014;8 (2):408–12. 10.1007/s12072-013-9489-1
43 Xu XL, Xing BC, Han HB, Zhao W, Hu MH, Xu ZL, et al. The properties of tumor-initiating cells from a hepatocellular carcinoma patient’s primary and recurrent tumor. Carcinogenesis. 2010;31(2):167–74. 10.1093/carcin/bgp232
44 Oliva J, French BA, Qing X, French SW. The identification of stem cells in human liver diseases and hepatocellular carcinoma. Exp Mol Pathol. 2010;88(3):331–40. 10.1016/j.yexmp.2010.01.003
45 Yin X, Li YW, Zhang BH, Ren ZG, Qiu SJ, Yi Y, Fan J. Coexpression of stemness factors Oct4 and Nanog predict liver resection. Ann Surg Oncol. 2012;19(9):2877–87. 10.1245/s10434-012-2314-6
46 Fan H, Cui Z, Zhang H, Mani SK, Diab A, Lefrancois L, et al. DNA demethylation induces SALL4 gene re-expression in subgroups of hepatocellular carcinoma associated with Hepatitis B or C virus infection. Oncogene. 2017;36(17):2435–45. 10.1038/onc.2016.399
47 Shin JG, Cheong HS, Lee K, Ju BG, Lee JH, Yu SJ, et al. Identification of novel OCT4 genetic variant associated with the risk of chronic hepatitis B in a Korean population. Liver Int. 2017;37(3):354–61. 10.1111/liv.13245
48 Sartorius K, Makarova J, Sartorius B, An P, Winkler C, Chuturgoon A, et al. The regulatory role of microrna in hepatitis-b virus-associated hepatocellular carcinoma (HBV-HCC) pathogenesis. cells. 2019;8(12). 10.3390/cells8121504
49 Bautista WW, Osiowy C, Klein J, Minuk GY. Hepatitis B Virus infection of normal hepatic stem/progenitor cells. J Clin Exp Hepatol. 2019;9(1):34–42. 10.1016/j.jceh.2018.02.002
50 Yang H, Mo J, Xiang Q, Zhao P, Song Y, Yang G, et al. SOX2 represses hepatitis B virus replication by binding to the viral enhii/cp and inhibiting the promoter activation. Viruses. 2020;12(3):273. 10.3390/v12030273